
Abstract
Background
To evaluate the safety, tolerability, pharmacokinetics, and dose-limiting toxicities of a single intravitreal (IVT) injection of PF-04523655, a 19-nucleotide, O-methyl stabilized, double-stranded small interfering ribonucleic acid targeting the RTP801 gene in patients with neovascular age-related macular degeneration (AMD).
Methods
Prospective, phase 1, clinical multicentre trial, enrolled 27 patients with neovascular AMD unresponsive to prior treatment and best corrected visual acuity (BCVA) ≤20/200 in the study eye in stratum 1: (dose-escalating, open-label: 50 to 3000 μg of PF-04523655) and 27 patients who had potential to benefit from therapy and BCVA of ≤20/100 and ≥20/800 in stratum 2 (parallel, masked study of 1000, 1500, 2250, and 3000 μg of PF-04523655). The primary outcome was safety and tolerability assessment as well as pharmacokinetic profiling following a single IVT injection of PF-04523655.
Results
Doses of PF-04523655 ≥400 μg were generally detectable in the plasma at 1, 4, and 24 h post-injection. And all doses were below the lowest level of quantification by day 14. A single IVT injection of 50 to 3000 μg of PF-045237655 was generally safe and well tolerated over 24 months. There were no dose-limiting toxicities.
Conclusion
A single IVT injection of PF-0523655 ≤3000 μg seems safe and well tolerated in eyes with neovascular AMD.
Similar content being viewed by others


Introduction
Neovascular age-related macular degeneration (AMD) occurs in 4 to 8% of the individuals over the age of 70 years.1, 2 Current interventions are mainly focused on vascular endothelial growth factor (VEGF) blockade to inhibit abnormal angiogenesis that leads to choroidal neovascularization (CNV) and eventual retinal neural apoptosis/death. Anti-VEGF therapies have been shown to stabilize disease progression and improve vision. However, as they act exclusively on blood vessel formation, their effect on restoration of vision is, at best, moderate.3, 4, 5, 6 Hence, there is a need for additional therapies that directly protect the retinal neurons within existing lesions and enhance survival of damaged neurons, thus promoting the restoration of visual acuity.
PF-04523655, a small interfering ribonucleic acid (siRNA) that acts via RNA interference (RNAi) to inhibit the expression of the hypoxia-inducible gene, RTP801, is being developed for the treatment of CNV secondary to AMD and diabetic retinopathy, including diabetic macular edema.7, 8, 9 Expression of the RTP801 gene is upregulated in response to hypoxia and/or oxidative stress, which leads to the induction of neuronal cell apoptosis.10, 11, 12, 13 The different mechanism of action of PF-04523655 (ie, blocking the RTP801 hypoxia/stress pathway) is independent of, and potentially complementary to, the anti-angiogenesis mechanism of the existing anti-VEGF therapies.
Unlike other siRNAs that have been evaluated for the treatment of neovascular AMD,14 PF-04523655 is a 19-nucleotide, methylated double-stranded siRNA specifically targeting the RTP801 gene. In addition to its inhibition of the RTP801 gene, the methylation, double stranding and short length of PF-04523655 may facilitate cellular entry and prevent its degradation in vivo.15, 16 These structural and chemical properties may make PF-04523655 more effective than other siRNAs for the treatment of neovascular AMD.
PF-04523655 was well tolerated without indications of systemic toxicity, following intravenous and/or intravitreal (IVT) administration for up to 4 weeks in rats and 13 weeks in cynomolgus monkeys. Following IVT injections in the monkeys at the highest dose level tested (3 mg/eye), there was an increased incidence of cellular infiltration, and elevated intraocular pressure (Pfizer data on file). There was no indication of potential genetic toxicity.
This first-in-human study was designed to evaluate the safety, tolerability, pharmacokinetics (PK), and dose-limiting toxicities of a single IVT injection of doses of PF-04523655 ranging from 50 to 3000 μg in patients with advanced neovascular AMD, in anticipation of initiating clinical studies of the safety and efficacy of PF-04523655 for the treatment of AMD and diabetic macular edema.
Materials and methods
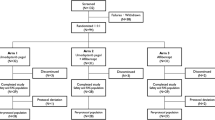
This trial was a phase 1, prospective, multicentre, dose-escalation study to evaluate the safety, tolerability, and dose-limiting toxicities and to establish the PK of PF-04523655 when administered as a single IVT in patients with CNV secondary to AMD. The study included a 2-week screening period, an 84-day study period (visits at days 0, 1, 7, 14, 28, 56, and 84), and a post-treatment follow-up of 24 months (visits at months 6, 9, 12, 18, and 24). The study was conducted in two strata. Stratum 1 was an open-label, dose-escalating safety study in patients unlikely to improve visual acuity or who failed previous treatment. Stratum 2, was a randomized, masked study in patients likely to improve visual acuity using the ≥1000 μg doses from stratum 1.
Patients were recruited at retinal specialty clinical sites in the United States of America and Israel. Consented patients of either gender ≥50 years of age, in general good health were enrolled. For stratum 1, patients were required to have a best corrected visual acuity (BCVA) ≤20/200 because of advanced neovascular AMD that, in the opinion of the investigator, was not likely to show significant improvement in BCVA from currently available treatment options (eg, ranibizumab, pegaptanib, bevacizumab, photodynamic, or steroid-based therapy). In addition, the patients must have failed to benefit from at least one of these treatment options.
In stratum 1, the single IVT injection dose of PF-04523655 was escalated as follows: 50, 100, 200, 400, 670, 1000, 1500, 2250 and 3000 μg. Dose escalation occurred when the dose-limiting toxicities were not observed during the 7 days following the previous dose cohort. Decisions regarding dose escalation were made only after consultation with the investigators and the safety advisory board.
After the initial safety of PF-04523655 was established in stratum 1 and after a safety report was submitted to the regulatory authorities, the sponsor decided to explore, in stratum 2, possible efficacy of the higher doses of PF-04523655: 1000, 1500, 2250 and 3000 μg. For stratum 2, patients were enrolled who, in the opinion of the investigator, had the potential to show improvement in BCVA from other treatment options and had a BCVA between 20/100 and 20/800 inclusive. Twelve patients were equally randomized to the four high doses of PF-04523655, and then an additional 15 patients were assigned to the 3000 μg dose group.
Patients already taking vitamin supplements and/or trace minerals for AMD at baseline were permitted to continue to do so. However, patients were asked not to begin new AMD treatments, such as vitamin supplements, trace minerals, or prescription medications, during the active phase of this study (the first 84 days). Patients were permitted to receive any AMD treatment as needed after the single PF-04523655 IVT injection; however, only data collected prior to the additional AMD treatment were included in the BCVA and the central retinal thickness analyses.
Safety and tolerability were the primary outcome measures for the entire 24-month study period. In stratum 1, plasma concentrations of PF-04523655 were measured at preinjection and 1, 4, 24, 168, and 336 h after injection for PK assessments. In stratum 2, BCVA and central retinal thickness measurements were assessed at screening, days 1, 7, 14, 28, 56, and 84.
BCVA using the early treatment for diabetic retinopathy study (EDTRS) protocol, intraocular pressure, biomicroscopy, funduscopy, and time-domain optical coherence tomography (Stratus OCT, Carl Zeiss Meditec Inc., Dublin, CA, USA) were performed at screening, days 1, 7, 14, 28, 56, and 84, and all follow-up visits. OCT, fundus photography and fluorescein angiography evaluations of the study eye through day 84 were read by an independent reading centre, the Digital Angiography Reading Center (DARC, New York, NY, USA).
There were no formal statistical tests of hypotheses. All statistical methods were descriptive only. All patients who received any study drug in stratum 1 or stratum 2 were included in the safety analysis. Stratum 1 patients with at least 1 detectable PF-04523655 plasma level were included in the PK analysis. In stratum 2, BCVA and central retinal thickness were evaluated through day 84. For patients who received additional AMD treatment, only data collected prior to the treatment were included in the analyses.
Pharmacokinetic parameters including maximum observed plasma drug concentration (Cmax), time to maximum observed drug concentration (Tmax), and area under the plasma concentration-time curve based on the last observed concentration (AUClast) were estimated using the standard non-compartmental method. For a patient, at least two concentrations above the lowest level of quantization (LLOQ) after IVT injection were required to calculate AUClast.
All patients read and signed an informed consent in accordance with Good Clinical Practices, the World health Organization Declaration of Helsinki 1996, and Health Insurance Portability and Accountability Act. The safety of the trial was assessed by an independent safety advisory board. The clinical trial was registered on the www.clinicaltrials.gov website, identifier NCT00725686.
Results
The study enrolled 27 patients in stratum 1 (n=3 in each of the 9 dose groups) and 27 patients in stratum 2 (n=3 for the 1000, 1500, and 2250 μg groups; n=18 for the 3000 μg group) at 11 retinal clinical sites. The patient demographics, including baseline mean BCVA and central retinal thickness, are given in Tables 1a and 1b. Twenty-six patients in stratum 1 and all 27 patients in stratum 2 completed day 84. The first patient, first visit was on 1 February 2007 and the last patient’s month 24 visit occurred on 29 November 2010. The mean changes in BCVA and central retinal thickness from baseline for stratum 2 are shown in Figures 1a and b.

(a) Mean change in BCVA from screening in Stratum 2 patients (excluding data collected after any additional AMD treatments). (b) Mean change in retinal thickness (optical coherence tomography graded by the DARC Reading Center) from screening in Stratum 2 patients (excluding data collected after any additional AMD treatments).
Dose-limiting toxicities were not observed in this study. In total, 20 (74.1%) patients in stratum 1 and 19 (70.4%) patients in stratum 2 reported at least one adverse event (AE). Most AEs were of mild or moderate severity and unrelated to study drug. Nineteen (35%) patients experienced at least one AE attributed to the IVT injection procedure. The most frequently reported AEs (n≥2) are presented in Tables 2 and 3.
During the follow-up period, three patients in stratum 2 died from causes unrelated to study treatment. Across both strata, there were three serious AEs reported (chronic obstructive pulmonary disease, atrial fibrillation, and hemolytic anemia) and all were considered unrelated to the study drug.
There were three reports of retinal pigment epithelial detachments occurring after the patients started receiving IVT injections of ranibizumab in the study eye. One of these three retinal pigment epithelial detachments was considered possibly related to the prior PF-04523655 (1500 μg) treatment.
PF-04523655 reached maximal concentrations in the plasma between 1 and 4 h after injection at all doses, except the 50 and 200 μg doses. Plasma levels of PF-04523655 were generally below LLOQ at doses ≤200 μg at all time points. Although the highest dose of PF-04523655 administered was 3000 μg, the highest median plasma concentration observed was 2.86 ng/ml at 4 h after administration of the 2250 μg dose. Plasma concentrations fell below the LLOQ by 168 h (7 days) for all doses, except 3000 μg, which fell below LLOQ by 336 h (14 days) after injection. The differences between the plasma levels of PF-04523655 after these doses may not be significant because of the small number of observations (n=3).
At doses greater than or equal to 400 μg, there was sufficient data to derive PK parameters; however, there was large variability in both the AUC and Cmax values. There appeared to be a dose-dependent (but not dose-proportional) increase in systemic exposure to PF-04523655 with increasing dose, although exposures were quite variable even within a given dose group (Table 4). There was insufficient information on plasma concentrations beyond 24 h after dose administration to determine the elimination half-life (t½).
Discussion
This multicentre phase 1 clinical trial of the first-in-human safety/tolerability and PK study following a single IVT injection of PF-04523655 at doses ranging from 50 to 3000 μg demonstrates PF-04523655 to be well tolerated without any significant safety issues. The majority of reported AEs were mild and moderate, and there was no evidence that AEs were related to systemic exposure. No dose-limiting toxicities were observed in this study and therefore a maximum tolerated dose was not established.
The PF-04523655 plasma concentrations after a single IVT injection were measurable at doses of ≥400 μg at 1, 4, and 24 h after the dose. Only one subject in the 3000 μg dose group had a measurable PF-04523655 concentration at 168 h (day 7) post-dose and no subject had a measurable PF-04523655 concentration at 336 h (day 14) post-dose. The lack of human plasma concentrations beyond 24 h precluded calculation of the elimination half-life (t½). This study did not evaluate metabolism or excretion of PF-04423665; however, it is expected that similar to other oligonucleotides, the nuclease-mediated metabolites of PF-04423655 would be excreted in the urine.
Although there appeared to be a dose-dependent increase in systemic exposure to PF-04523655, it was not dose-proportional. Systemic exposures were quite variable across dose groups. Even within a dose group, the three patients who received the same dose yielded quite variable PK parameters. There was no evidence that the highest systemic exposures of 40 to 65 ng/h/ml were associated with the occurrence of more frequent AEs. In this limited, small, uncontrolled, open-label, phase 1 trial, the sample size was not sufficient to statistically evaluate efficacy. The preliminary safety of a single PF-04523655 IVT injection supports further dose-ranging studies of monthly IVT injections of PF-04523655.

References
Klein R, Klein BEK, Linton KLP . Prevalence of age-related maculopathy: The Beaver Dam eye study. Ophthalmology 1992; 99: 933–943.
Jonasson F, Arnarsson A, Eirikadottir G, Harris TB, Launer LJ, Meuer SM et al. Prevalence of age-related macular degeneration in old persons: age, gene/environment susceptibility Reykjavik Study. Ophthalmology 2011; 118: 825–830.
Martin DF, Maguire MG, Ying GS, Grunwald JE, Fine SL, Jaffe GJ, CATT Research Group. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med 2011; 364: 1897–1908.
Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, Kaiser PK, Chung CY et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med 2006; 355: 1419–1431.
Avery RL, Pieramici DJ, Rabena MD, Castellarin AA, Nasir MA, Giust MJ . Intravitreal bevacizumab (Avastin) for neovascular age-related macular degeneration. Ophthalmology 2006; 113: 363–372.
Heier JS, Boyer D, Nguyen QD, Marcus D, Roth DB, Yancopoulos G et al. The 1-year results of CLEAR-IT 2, a phase 2 study of vascular endothelial growth factor trap-eye dosed as-needed after 12-week fixed dosing. Ophthalmology 2011; 118: 1098–1106.
Brafman A, Mett I, Shafir M, Gottlieb H, Damari G, Gozlan-Kelner S et al. Inhibition of oxygen-induced retinopathy RTP801-deficient mice. Invest Ophthalmol Vis Sci 2004; 45: 3796–3805.
Rittenhouse KD, Hirakawa B, Huang W, Basile AS, Johnson TR, Schachar RA . Dose-related gene silencing of RTP801 with the siRNA PF04523655 in Long Evans rat models of STZ induced diabetes and laser induced CNV. Invest Ophthalmol Vis Sci 2010; 51, E-Abstract 6447.
Rittenhouse KD, Kalabat D, Yang A, Vicini P, Johnson TR, Huang W et al. Characterization of regional RTP801 gene expression within the retina and the concentration-effect relationship of PF-655, an RTP801-silencing siRNA, following intravitreous administration to diabetic rats. Invest Ophthalmol Vis Sci 2011; 52, E-Abstract 5641.
Shoshani T, Faerman A, Mett I, Zelin E, Tenne Y, Gorodin S et al. Identification of a novel hypoxia-inducible factor1-responsive gene, RTP801, involved in apoptosis. Mol Cell Biol 2002; 22: 2283–2293.
Jin H-O, An S, Lee H-C, Woo SH, Seo SK, Choe TB et al. Hypoxic condition-and high cell density-induced expression of Redd1 is regulated by activation of hypoxic-inducible factor-1a and Sp1 through the phosphatidylinositol 3 kinase/Akt signaling pathway. Cell Signal 2007; 19: 1393–1403.
Ellisen LW, Ramsayer KD, Johannessen CM, Yang A, Beppu H, Minda K et al. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol Cell 2002; 10: 995–1005.
Feinstein E, Ashush H, Kleinman ME, Nozaki M, Kalinski H, Mett I et al. PF-04523655 (REDD14), an siRNA compound targeting RTP801, penetrates retinal cells producing target gene knockdown and avoiding TLR3 activation. Invest Ophthalmol Vi Sci 2009; 50, E-Abstract 5693.
Kaiser PK, Symons RCA, Shah SM, Quinlan EJ, Tabandeh H, Do DV et al. RNAi-based treatment for neovascular age-related macular degeneration by Sirna-027. Am J Opthalmol 2010; 150: 33–39.
Whitehead KA, Langer R, Anderson DG . Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov 2009; 8: 129–138.
Perkel JM . RNAi therapeutics: a two-year update. Science 2009; 326: 454–456.
Acknowledgements
This study was supported by Pfizer Inc. San Diego, California, USA.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
RA Schachar, CI Nduaka, M Sperling, AS Basile, K Chi-Burris, KJ Klamerus, E Yan, DA Paggiarino were employees of Pfizer at the time of the study; QD Nguyen and I Rosenblatt were investigators; and A Khan, R Aitchison, and SS Erlich were employees of Quark Phamaceuticals Inc. at the time of the study.
Appendix
Appendix
Tom S Chang, Retina Institute of California, Pasadena, CA, USA; Michaella Goldstein, Tel Aviv Sourasky Medical Center, Tel Aviv, Israel; Peter Kaiser, Cole Eye Institute, Cleveland, OH, USA; James M Klancnik, Vitreous-Retina-Macula, Consultants of New York, New York, NY, USA; Gregg Kokame, Retina Consultants of Hawaii, Aiea, Hawaii; Quan Dong Nguyen, Wilmer Eye Institute, Baltimore, MD, USA; Roger L Novack, Retina-Vitreous Associates Medical Group, Beverly Hills, CA, USA; Anat Pilpul, Kaplan Medical Center, Rehovot, Israel; Irit Rosenblatt, Rabin Medical Center, Petah Tikva, Israel; Philip J Rosenfeld, Bascom Palmer Eye Institute, Miami, FL, USA; Daniel Ting, Bay Area Retina Associates, Walnut Creek, CA, USA.
Rights and permissions
About this article
Cite this article
Nguyen, Q., Schachar, R., Nduaka, C. et al. Phase 1 dose-escalation study of a siRNA targeting the RTP801 gene in age-related macular degeneration patients. Eye 26, 1099–1105 (2012). https://doi.org/10.1038/eye.2012.106
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2012.106
Keywords
This article is cited by
-
The stress-responsive protein REDD1 and its pathophysiological functions
Experimental & Molecular Medicine (2023)
-
Expression profiles and prognostic value of miRNAs in retinoblastoma
Journal of Cancer Research and Clinical Oncology (2019)
-
siRNA Versus miRNA as Therapeutics for Gene Silencing
Molecular Therapy - Nucleic Acids (2015)
-
Multigenic lentiviral vectors for combined and tissue-specific expression of miRNA- and protein-based antiangiogenic factors
Molecular Therapy - Methods & Clinical Development (2015)
-
CAPN5 gene silencing by short hairpin RNA interference
BMC Research Notes (2014)
