
Abstract
Aims
The aim of this study is to provide a clinical update on optic neuritis (ON), its association with multiple sclerosis (MS), and neuromyelitis optica (NMO).
Methods
This study included a PubMed review of the literature written in the English language.
Results
ON in adults is typically idiopathic or demyelinating, and is characterised by unilateral, subacute, painful loss of vision that is not associated with any systemic or other neurological symptoms. Demyelinating ON is associated with MS, and we review the key studies of ON including the ON treatment trial and several other MS treatment trials and NMO.
Conclusion
Acute demyelinating ON can occur in isolation or be associated with MS. Typical ON does not require additional evaluation other than cranial magnetic resonance imaging. NMO is likely a separate disorder from MS and the ON in NMO has a different treatment and prognosis.
Methodology
The authors conducted an English language search using Pubmed from the years 1964 to 2010 using the search terms ‘ON’, ‘MS’ and ‘NMO’. The authors included original articles, review articles, and case reports, which revealed new aspects as far as epidemiology, histopathology, clinical manifestations, imaging, genetics, and treatment of ON. Titles were reviewed for topicality and full references were obtained. Letters to the editor, unpublished work, and abstracts were not included in this review.
Similar content being viewed by others



Introduction
We use the term, optic neuritis (ON) to define an acute, demyelinating, or idiopathic optic neuropathy. ON can present in isolation or be associated with multiple sclerosis (MS) or occur in the setting of neuromyelitis optica (NMO). Although secondary causes of ON include infectious and inflammatory aetiologies other than MS and NMO, we emphasise demyelinating ON in this review. ON of the type associated with MS (even where there is no evidence of MS at the time of presentation) is sometimes referred to as ‘typical’ although, as described below, what could be regarded as typical in one part of the world may not be in another. It is also important to recognise that, although demyelination is a feature of ON in both MS and NMO, it is not the sole pathological feature; there is evidence of axonal loss in both conditions (see below) and of astrocytic damage in NMO.1
Several clinical trials, including the ON treatment trial (ONTT), have provided useful data on the management of ON. This article will review our current understanding of ON, its association with MS, some relevant MS clinical trials, and NMO. It is worth noting that this review is based on the Western, English language, literature, and the reader should recognise that there are world-wide differences in the prevalence and presentation of MS as well as differences in the aetiologies of inflammatory optic neuropathies and that the evaluation and management of ON might differ depending on the epidemiology of the local population being studied.
Pathophysiology
The presumed pathophysiology of ON is inflammation and demyelination of the optic nerve. Activated peripheral T cells migrate across the blood–brain barrier and release cytokines and other inflammatory mediators leading to neuronal cell death and axonal degeneration.2 Although multiple studies in MS3, 4, 5, 6 have demonstrated inflammatory demyelination as the pathological hallmark of disease; after the acute event, axonal damage leading to axonal loss can lead to severe and sometimes irreversible neurological impairment.7
Optical coherence tomography (OCT) has provided one objective means to quantify this axonal loss. In one study, 74% of patients with acute ON demonstrated retinal nerve fibre layer (RNFL) thinning within 3 months on OCT.8 One study looked at cases selected to investigate the full range of visual outcomes seen in ON (with, therefore, a higher proportion of cases with severe visual loss than would be seen in an unselected cohort) and found a direct correlation between the degree of RNFL thinning on OCT and the final level of visual impairment,9 although other studies have not shown as convincing a correlation. Thus, in addition to reducing the number and severity of attacks, preventing axonal loss and subsequent disability is an additional goal of many of the existing and emerging therapies for both ON and for MS.
Epidemiology
The incidence of ON is highest in populations located at higher latitudes (eg, Northern United States; Northern and Western Europe; New Zealand and Southern Australia) compared with geographic locations closer to the equator.10, 11 In the United States of America, studies have estimated the annual incidence of ON as 5 to 6.4 per 100 000, with a prevalence of 115 per 100 000.12, 13 The demographics for ON would be expected to closely follow those seen in MS, although there is no data to support this known to the authors. ON is seen more commonly in Caucasians, and is uncommon but reported in black populations. Whites of Northern European descent develop ON eight times more frequently than blacks and Asians.14, 15, 16 In Asia, ON is proportionately more common relative to the incidence of MS in the United States of America or Western Europe.17 ON is less frequent in South America and in the Mediterranean region but newer studies have reported an increasing prevalence in the last few decades.18, 19, 20
Some studies have shown that individuals who migrate before puberty have the incidence of MS in the area to which they have migrated.20, 21, 22 This suggests a possible interaction between the ethnic origin of the patient, the latitude, and whatever is the factor (or factors) that triggers MS. Other studies have suggested that less sun exposure at increasing latitude might be a risk factor for MS,23, 24 and vitamin D deficiency is an additional potential explanation for some of the epidemiological differences in MS.25, 26 In addition, an increased incidence of ON during the spring season has been reported.27, 28 There are also some who postulate viruses as having a role in the pathogenesis of MS.29, 30
Diagnosis
Clinical presentation (signs and symptoms)
Patients with acute demyelinating ON are typically healthy young adults and most patients present between 20 and 45 years of age. ON can be subclassified into four categories on the basis of site of involvement:
-
1
Retrobulbar neuritis: normal disc appearance.
-
2
Papillitis: a swollen optic disc (Figure 1).
Figure 1 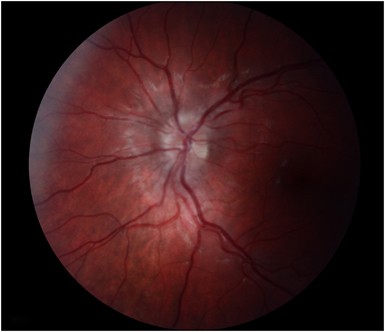
A 14-year-old male with papillitis.
-
3
Perineuritis: involvement of the optic nerve sheath rather than the optic nerve parenchyma per se (seen radiographically). The optic disc in this setting may or may not be swollen.
-
4
Neuroretinitis: optic disc oedema and a ‘star figure’ of macular exudates.

Retrobulbar neuritis and papillitis are most often associated with MS. Perineuritis and neuroretinitis are distinct in their tissue of involvement and are more often associated with an infectious (eg, cat scratch disease, syphilis) or inflammatory (eg, sarcoidosis) aetiology rather than MS. We will not be discussing in detail the latter two entities in this article.
There is a female preponderance by a ratio of 3 : 1.31 Demyelinating ON associated with MS is less common in children than in adults, and in children ON is often related to a post- or para-infectious demyelination. Children will more often present with bilateral simultaneous involvement (60–70% of cases) and more profound visual loss initially.32, 33, 34, 35
The classic presentation of ON is unilateral loss of vision in the majority of adults. The vision loss is quite variable and ranges from mild to no light perception. Typically there is a short period of progression over hours to 10 days followed by improvement: complete or almost complete recovery of visual acuity and visual field are common.36, 37 Progressive visual worsening for more than 2 weeks, or lack of recovery after 8 weeks, should suggest an alternative diagnosis to demyelinating (MS type) ON.38 Periocular pain and retro-orbital pain occurs in >90% of cases, usually exacerbated by eye movement, may precede or coincide with the visual symptoms, and usually resolve over days. Most patients show reduced contrast sensitivity and dyschromatopsia, which are often out of proportion to the visual acuity deficit. When colour loss is present, most persons show mixed red–green and blue–yellow colour defects.39 Up to 30% of patients experience positive visual phenomena (photopsias), which may occur at the onset of visual symptoms or during the course of the disorder.31
Any type of nerve-fibre-bundle-related visual field defect is possible in ON. In the 15 year follow-up report from the ONTT, diffuse and central loss predominated in the affected eye at the initial visit then most commonly, partial arcuate, paracentral, and arcuate type of nerve fibre bundle-type visual field defects during follow-up40 (Figure 2a). The pattern of visual field loss in ON is of limited value in distinguishing ON from other optic neuropathies but hemianopic visual field defects are uncommon and should raise suspicion for a compressive or alternate aetiology to ON. We recommend that a formal visual field be carried out in patients with ON, and that visual fields be used to follow patients for improvement over time (Figure 2b). In unilateral (or in asymmetric but bilateral cases) a relative afferent pupillary defect (RAPD) is found. In bilateral and symmetric cases or in cases with a preexisting optic neuropathy (eg, before ON—which may be subclinical) in the fellow eye, an RAPD may not be apparent.31, 41 A neutral density filter could also be used to bring out a subtle RAPD,42 as well as to quantify the RAPD.

(a) A 33-year-old female with superior arcuate defect on visual field secondary to optic neuritis. (b) The same patient approximately at 4 months later after treatment with intravenous steroids.
About two-thirds of cases of acute demyelinating ON are retrobulbar and the optic disc appears normal. When present the disc swelling of demyelinating ON typically is diffuse31 (Figure 1), the presence of segmental changes, altitudinal swelling, pallor, arteriolar attenuation, and splinter haemorrhages plus a small cup to disc ratio (ie, ‘disc at risk’) in the contralateral eye is more characteristic of non-arteritic anterior ischaemic optic neuropathy (NAION), but sometimes there is overlap in the clinical presentations of NAION and ON.31, 43
After ON resolves the disc often develops optic atrophy most commonly in the temporal aspect (Figure 3). Additional clinical findings of residual decreased subjective relative light intensity; contrast loss and dyschromatopsia; exercise- or heat-induced exacerbation of visual symptoms (Uhthoff phenomenon) can be seen,12, 13, 31, 44 and the Pulfrich phenomenon (anomalous perception of the direction of movement of an object occurs because of the asymmetry of conduction velocity in the optic nerves) can occur in patients after recovery from ON. ON has been shown to recur in the same eye or the contralateral eye. On the basis of ONTT, the 5- and 10-year risk for recurrence are 28 and 35%, respectively.37, 45 Recurrence rates are higher in those with MS.45

A 33-year-old female with optic atrophy secondary to optic neuritis.
Diagnostic evaluation, imaging, and laboratory evaluation
The diagnosis of ON is a clinical one.46, 47, 48 Patients with typical ON, according to the ONTT, do require neither laboratory studies nor a lumbar puncture (LP) for diagnostic purposes. Magnetic resonance imaging (MRI) of the brain with and without gadolinium is typically recommended for prognostic purposes for MS but at present does not influence treatment in the United Kingdom, but may when disease modifying treatments are commenced in patients with clinically isolated syndromes such as ON (see below). In the ONTT an abnormal baseline brain MRI was a strong predictor of MS after isolated ON in adults.45, 49
Contrast enhancement of the optic nerve is a sensitive finding in acute ON, reported in up to 94% of cases45 (Figure 4). In addition, thin (2–3 mm) section, fat-suppressed T2-weighted images, such as short τ-inversion recovery sequences, may show characteristic high-signal intensity in ON. ON enhancing lesions involving the canal or involving longer segments of optic nerve may have worse initial vision, but the location and length of enhancement are not predictive of recovery.50 There is speculation as to whether there are certain features on MRI in patients with acute ON that suggest NMO rather than MS, however there is no published data at present to assist in this distinction. Although visual evoked potentials (VEPs) may be helpful in detecting contralateral but subclinical cases of ON, in general, the use of VEP is not necessary for the diagnosis of ON.

A 37-year-old female with left optic nerve enhancement on magnetic resonance imaging consistent with optic neuritis.
Cerebrospinal fluid (CSF) analysis is usually not necessary in patients with typical demyelinating ON.51 In the ONTT, only the presence of oligoclonal bands correlated with later development of MS, but an abnormal baseline MRI was a better predictor of MS.51, 52 When performed, CSF analysis for cell count, protein, and immunoglobulin G (IgG) synthesis, IgG ratio, myelin basic protein, IgG κ-light chains, and oligoclonal banding might be useful for supporting a clinical diagnosis of MS in patients with typical ON. Patients with atypical ON might require a LP to exclude an alternative aetiology for an optic neuropathy however.
Acute treatment for ON
In the ONTT individuals randomised to placebo reached the same visual outcome at 6 months as those receiving steroid treatment. At 15 years later, visual acuity remained stable in most patients.53 Treatment with intravenous methylprednisolone (IVMP) resulted in a more rapid rate of visual recovery and may be important in monocular patients, patients with significant bilateral visual loss, or those with occupations requiring faster recovery to normal visual acuity. Treatment with oral prednisone alone in standard doses increased the recurrence rates of ON (30% at 2 years with oral prednisone vs 16% with placebo and 13% with IVMP) and is therefore not recommended in acute typical ON. However, a counter argument has been adopted by some authors10, 54 in view of the fact that the 95% confidence intervals (95% CI) for the relative risk ratio was wide, approaching unity, and at 10 years this difference remained a trend but was no longer statistically significant.45 Furthermore other studies using higher doses of oral corticosteroids have not shown any effect on the recurrence rate of ON compared with placebo.55 Whether the finding in the ONTT is related to the lower dose that was used, or to other factors, is currently unknown.
When prescribing steroid treatment, it is our usual and customary practise to inform patients of the common adverse effects of corticosteroids. In the ONTT, mild side effects56 of steroids included sleep disturbances, mild mood changes, stomach upset, and facial flushing. Serious side effects were rare and only occurred in those treated with IVMP. One individual developed severe depression that required psychotropic medications and another individual had acute pancreatitis that fully resolved without sequelae. There were no cases of peptic ulceration or haemorrhages. Avascular necrosis of the hip or other joints is a serious complication that has been reported to occur rarely as a result of a brief course of corticosteroids.57
Intravenous immunoglobulin (IVIG) and plasma exchange have been considered in the treatment of acute ON but have yielded little to no beneficial effect.58, 59, 60
In the ONTT treatment with IVMP followed by oral prednisone resulted in lower rates of MS development in the first 2 years,61 but this effect was not sustained after year 3 (7.5% for IVMP vs 14.7% for oral prednisone alone and 16.7% in the placebo group at 3 years). We do not use this secondary outcome measure of the ONTT as a rationale for using IV steroids in ON.
The bottom line in MS-associated ON is that there is little to be gained or lost by treating or not treating with corticosteroids in the acute situation. Of greater importance is the need to identify cases not due to MS, such as NMO, in which treatment is crucial to prevent irreversible visual loss. Criteria and algorithms to assist in the identification of atypical cases have been published.62, 63
Utility of MRI imaging
Cranial MRI imaging provides prognostic information for patients with ON for developing MS based on the number of demyelinating white matter lesions. The diagnosis of MS can be made either by dissemination of lesions in time and space based on clinical episodes (relapses) or by radiological grounds alone.64 All MRI studies have related the imaging findings to the risk of a second (MS defining) episode occurring within a chosen time interval. The longer the observation period the higher will be the proportion of cases achieving a clinical diagnosis of MS. In the absence of any lesions on a baseline brain MRI, patients with ON have a 25% cumulative probability of developing MS at 15 years. With the presence of one lesion, the 15-year cumulative probability of developing MS was 60% (HR, 2.80; 95% CI, 1.68–4.68); two lesions 68% (HR, 2.86; 95% CI, 1.43–5.00); three lesions 78% (HR, 4.46; 95% CI, 2.99–6.65)49 (Figure 5a–f). Thus the MRI is the single best predictor for MS in patients with ON. On the basis of the ONTT, however, the ophthalmologist counselling a patient with ON should be aware of the following: (1) ON with a normal MRI still carries a 15-year risk for MS of 25%, and therefore patients with a normal MRI at onset should not be told that they ‘do not have MS’, and conversely (2) a patient with ON with even three or more lesions on MRI have a 78% risk of MS should not be told that they ‘definitely have MS.’ More recent studies have shown that the sensitivity of imaging findings can be increased by the use of contrast medium and by repeating the scan after an interval of a few months to look for new lesions.65 Whatever the sensitivity of imaging in predicting a second episode, there will always be ‘false-negative’ and ‘false-positive’ findings.

(a–f) A 23-year-old female with hyperintense lesions on T2-weighted imaging (a–d) and post-gadolinium enhancing lesions T1-FLAIR imaging (e and f), consistent with demyelinating disease.
Immunomodulatory therapy
Immunomodulatory agents such as interferon β-1a (Avonex (Biogen Idec, Research Triangle Park, NC, USA), Rebif (EMD Serono, Inc., Rockland, MA, USA)), interferon β-1b (Betaseron, Bayer, Emeryville, CA, USA) and glatirimer acetate (Copaxone, Teva, Petach Tikva, Israel) have been referred to as ‘disease-modifying agents’ because of their role in increasing the time to onset of the second episode, and the frequency of subsequent MS relapses and demyelinating lesions, whether clinical or radiologically defined. Multiple, randomised, double-masked, placebo-controlled trials have looked at the utility of these immunomodulatory agents in patients with a clinically isolated syndrome (eg, ON) and at least two or more white matter lesions on brain MRI. Three such trials are the Controlled High-Risk Subjects Avonex Multiple Sclerosis Prevention Study,66 the Early Treatment of Multiple Sclerosis trial,67 and the Betaferon in Newly Emerging Multiple Sclerosis for Initial Treatment trial,68 and all demonstrated that interferon β prolonged the time interval to clinically definite MS. Treated patients were also found to have fewer lesions on brain MRI compared with placebo. Before committing a patient to treatment with interferon, it is important to keep in mind that over 40% of patients with ON and an abnormal MRI scan will not progress to CDMS at 10 years.69 In addition, patients will need to undergo treatment for approximately 6 years to prevent one relapse.67 The decision to start immunomodulatory therapy in patients with ON should be made by, or in conjunction with, a neurologist. We recommend that patients with ON undergo a cranial MRI. In the United States of America, if no lesions are present then many authors discuss with the patient the options of referral to a neurologist or a follow-up MRI in 6–9 months. If new lesions appear on the follow-up of MRI, even in the absence of new clinical symptoms or signs of MS, then a referral to neurology for discussion of MS treatment options can occur at that time. In patients with ON and demyelinating white matter lesions at onset in the United States of America, we recommend referral to a neurologist and a discussion about immunomodulatory therapy. On the other hand, for patients presenting with clinically isolated ON (or any other clinically isolated syndrome) in the United Kingdom, the policy remains that interferon β or Copaxone are not commenced unless a second clinical episode has occurred within 2 years of the episode of ON. This policy avoids the unnecessary treatment of patients who have infrequent episodes (on average less than one per year) and indeed a small number who untreated would not have a second relapse for several years or perhaps never.70
LP or spine cord imaging might be used to support the decision for treatment of MS. These ancillary investigations are of most value where the ON may be caused by a disorder other than MS.
Neuromyelitis optica
The ON in NMO (Devic disease), can resemble ON from MS initially, but NMO is a distinct demyelinating condition from MS (see Table 1). Patients can present with ON or transverse myelitis (TM) sequentially or simultaneously. The incidence and prevalence of NMO are largely unknown because of its under-recognition in the past. Approximately two-thirds of the patients are female. In North America, the median age of onset is in the fourth decade, which is about 10 years later than the typical age of onset in MS.71, 72 There appears to be a higher prevalence of NMO in certain non-Caucasian groups, such as African–Americans, South Asians, and Japanese.
Patients can present with unilateral or bilateral ON, TM or both in close temporal relationship to each other. TM manifests as deficiencies in both motor and sensory pathways as well as bowel and bladder dysfunction. Recognition of NMO is important because NMO generally carries a more aggressive and debilitating course compared with MS. At least 85% of patients will experience relapsing disease in the form of repeat episodes of ON, TM, or both.71, 72 Approximately 55% of relapses occur in the first year, 78% of relapses within 3 years, and 90% of relapses within 5 years.71 As opposed to typical ON patients with ON in NMO have a higher likelihood of permanent visual loss. Likewise for TM in NMO, in up to 30% of cases permanent paralysis in one or both legs can result. Approximately 50% of individuals with relapsing disease will experience paralysis or blindness within 5 years. Those patients with TM in the cervical cord can occasionally experience respiratory failure and even death.71
The diagnosis of NMO requires both clinical and radiographic findings.73 In addition to a LP and brain MRI, MRI imaging of the spine is essential in the workup for NMO. The diagnosis of NMO requires two absolute criteria and two of the three supportive criteria73 (see Table 2). Absolute criteria are ON and acute myelitis. The supportive criteria include brain MRI not meeting diagnostic criteria for MS,74 spinal cord MRI that has T2 signal abnormalities extending over three or more vertebral segments, or NMO–IgG seropositive status.
On histological sections of the spinal cord of NMO patients, findings include perivascular inflammatory demyelination to necrosis of both grey and white matter. A higher proportion of polymorphonuclear cells and eosinophils are apparent in the inflammatory reaction, a distinctive feature of NMO.75 A useful serum marker for NMO is the presence of NMO–IgG, which is believed to cause demyelination, both in NMO and in isolated ON.76 NMO–IgG is an antibody that targets aquaporin-4, a cellular transmembrane water channel.77, 78 The sensitivity and specificity of NMO–IgG is reportedly 76 and 94%.73
The first-line treatment for NMO is corticosteroids. If symptoms fail to improve, plasmapheresis may be considered. In a randomised, double-masked, sham-controlled study looking at the utility of plasmapheresis in patients with idiopathic inflammatory demyelinating diseases (including NMO), 42.1 vs 5.9% of patients experienced moderate or greater improvement in neurological function.79 Hospital admission may be necessary for patients with severe TM in order to closely monitor for any complications such as thromboembolic events or infections such as pneumonia or urinary tract infections. Immunomodulatory therapies, which are helpful in MS, are not the treatment of choice for NMO and in general, immunosuppressive rather than immunomodulatory therapy is recommended in NMO. Other therapies include mycophenolate mofetil, azathioprine, mitoxantrone, cyclophosphamide, and IVIG, all of which have been tried in NMO with varying success. A case series describing the use of rituximab in patients with NMO has shown promising results.80 We recommend testing for NMO in atypical ON, in patients with TM, or in patients with unexplained bilateral simultaneous or sequential ON especially if the cranial MR is not diagnostic for MS or if the spinal fluid shows marked pleocytosis (>50 cells).81
Conclusion
ON is the most common cause of acute optic neuropathy in young patients. In most cases, visual recovery will occur spontaneously but i.v. steroids can speed the rate of recovery. There is an association of ON with MS and baseline MRI imaging provides important prognostic information for MS. Typical ON can usually be diagnosed clinically, and according to the ONTT does not require routine laboratory or CSF testing, however atypical ON may require further tests to rule out secondary causes of ON. Some patients with atypical, bilateral, or unrecovered ON and patients with TM should be considered for NMO antibody testing. The treatment and prognosis of MS and NMO are different and a high index of suspicion should be maintained for atypical cases of ON.
References
Takano R, Misu T, Takahashi T, Sato S, Fujihara K, Itoyama Y . Astrocytic damage is far more severe than demyelination in NMO: a clinical CSF biomarker study. Neurology 2010; 75: 208–216.
Söderström M, Link H, Sun JB, Fredrikson S, Wang ZY, Huang WX . Autoimmune T cell repertoire in optic neuritis and multiple sclerosis: T cells recognising multiple myelin proteins are accumulated in cerebrospinal fluid. J Neurol Neurosurg Psychiatry 1994; 57: 544–551.
Prineas J . Pathology of multiple sclerosis. In: Cook S (ed.). Handbook of Multiple Sclerosis. Marcel Dekker: Basel (Switzerland), 2001, pp 289–324.
Trapp BD, Ransohoff RM, Fisher E, Rudick RA . Neurodegeneration in multiple sclerosis: relationship to neurological disability. Neuroscientist 1999; 5: 48–57.
Bjartmar C, Wujek JR, Trapp BD . Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease. J Neurol Sci 2003; 206: 165–171.
Brück W, Stadelmann C . Inflammation and degeneration in multiple sclerosis. Neurol Sci 2003; 24: S265–S267.
Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L . Axonal transaction in the lesions of multiple sclerosis. N Engl J Med 1998; 338: 278–285.
Costello F, Coupland S, Hodge W, Lorello GR, Koroluk J, Pan YI et al. Quantifying axonal loss after optic neuritis with optic coherence tomography. Ann Neurol 2006; 59: 963–969.
Trip SA, Schlottmann PG, Jones SJ, Altmann DR, Garway-Heath DF, Thompson AJ et al. Retinal nerve fiber layer axonal loss and visual dysfunction in optic neuritis. Ann Neurol 2005; 58: 383–391.
Shams PN, Plant GT . Optic neuritis: a review. Int MS J 2009; 16: 82–89.
Kurtzke JF . Optic neuritis or multiple sclerosis. Arch Neurol 1985; 42: 704–710.
Rodriguez M, Siva A, Cross SA, O’Brien PC, Kurland LT . Optic neuritis: a population based study in Olmsted County, Minnesota. Neurology 1995; 45: 244–250.
Percy AK, Nobrega FT, Kurland LT . Optic neuritis and multiple sclerosis. An epidemiologic study. Arch Ophthalmol 1972; 87: 135.
Phillips PH, Newman NJ, Lynn MJ . Optic neuritis in African Americans. Arch Neurol 1998; 55: 186–192.
Bhigjee AI, Moodley K, Ramkissoon K . Multiple sclerosis in KwaZulu Natal, South Africa: an epidemiological and clinical study. Mult Scler 2007; 13: 1095–1099.
Mbonda E, Larnaout A, Maertens A, Appel B, Lowenthal A, Mbede J et al. Multiple sclerosis in a black Cameronian woman. Acta Neurol Belg 1990; 90: 218–222.
Wakakura M, Minei-Higa R, Oono S, Matsui Y, Tabuchi A, Kani K et al. Baseline features of idiopathic optic neuritis as determined by a multicenter treatment trial in Japan. Optic Neuritis Treatment Trial Multicenter Cooperative Research Group (ONMRG). Jpn J Ophthalmol 1999; 43: 127–132.
El-Salem K, Al-Shimmery E, Horany K, Al-Refai A, Al-Hayk K, Khader Y . Multiple sclerosis in Jordan: a clinical and epidemiological study. J Neurol 2006; 253: 1210–1216.
Al-Araji A, Mohammed AI . Multiple sclerosis in Iraq: does it have the same features encountered in western countries? J Neurol Sci 2005; 15: 67–71.
Cristiano E, Patrucco L, Rojas JI . A systematic review of the epidemiology of multiple sclerosis in South America. Eur J Neurol 2008; 15: 1273–1278.
Kurtzke JF . Multiple sclerosis among immigrants. Br Med J 1976; 1: 1527–1528.
Dean G, Elian M . Age at immigration to England of Asian and Caribbean immigrants and the risk of developing multiple sclerosis. J Neurol Neurosurg Psychiatry 1997; 63: 565–568.
Cabre P . Environmental changes and epidemiology of multiple sclerosis in the French West Indies. J Neurol Sci 2009; 286: 58–61.
Cabre P, Signate A, Olindo S, Merle H, Caparros-Lefebvre D, Béra O et al. Role of return migration in the emergence of multiple sclerosis in the French West Indies. Brain 2005; 128: 2899–2910.
Zhang X, Wang W, Wei W, Wang Q, Wei Y, Kermode AG . Etiological profile of presumptive optic neuritis in China. J Clin Neurosci 2008; 15: 1346–1349.
Lim SA, Goh KY, Tow S, Fu E, Wong TY, Seah A et al. The pattern of ON as seen in Singapore differs from that reported in Caucasian studies and from the seminal Optic Neuritis Treatment Trial. Singapore Med J 2008; 49: 667–671.
Jin Y, de Pedro-Cuesta J, Söderström M, Stawiarz L, Link H . Seasonal patterns in optic neuritis and multiple sclerosis: a meta-analysis. J Neurol Sci 2000; 181: 56–64.
Balashov KE, Pal G, Rosenberg ML . Optic neuritis incidence is increased in spring months in patients with asymptomatic demyelinating lesions. Mult Scler 2010; 16: 252–254.
Allen L, Brankin B . Pathogenesis of multiple sclerosis—the immune diathesis and the role of viruses. J Neuropathol Exp Neurol 1993; 52: 95–105.
Power C, Antony JM, Ellestad KK, Deslauriers A, Bhat R, Noorbakhsh F . The human microbiome in multiple sclerosis: pathogenic or protective constituents? Can J Neurol Sci 2010; 37: S24–S33.
Balcer LJ . Clinical practice. Optic neuritis. N Engl J Med 2006; 354: 1273–1280.
Boomer JA, Siatkowski RM . Optic neuritis in adults and children. Semin Ophthalmol 2003; 18: 174–180.
Brady KM, Brar AS, Lee AG, Coats DK, Paysse EA, Steinkuller PG . Optic neuritis in children: clinical features and visual outcome. J Amer Acad Pediatr Ophthlamol Strab 1999; 3: 98–103.
Morales DS, Siatkowski RM, Howard CW, Warman R . Optic neuritis in children. J Pediatr Ophthalmol Strabismus 2000; 37: 254–259.
Lucchinetti CF, Kiers L, O’Duffy A, Gomez MR, Cross S, Leavitt JA et al. Risk factors for developing multiple sclerosis after childhood optic neuritis. Neurology 1997; 49: 1413–1418.
Celesia GG, Kaufman DI, Brigell M, Toleikis S, Kokinakis D, Lorance R et al. Optic neuritis: a prospective study. Neurology 1990; 40: 919–923.
Optic Neuritis Study Group. Visual function 5 years after optic neuritis: experience of the Optic Neuritis Treatment Trial. Arch Ophthalmol 1997; 115: 1545–1552.
Kaur P, Bennett JL . Optic neuritis and the neuro-ophthalmology of multiple sclerosis. Int Rev Neurobiol 2007; 79: 633–663.
Schneck ME, Haegerstrom-Portnoy G . Color vision defect type and spatial vision in the optic neuritis treatment trial. Invest Ophthalmol Vis Sci 1997; 38: 2278–2289.
Keltner JL, Johnson CA, Cello KE, Dontchev M, Gal RL, Beck RW et al. Visual field profile of optic neuritis: a final follow-up report from the optic neuritis treatment trial from baseline through 15 years. Arch Ophthalmol 2010; 128: 330–337.
Cox TA, Thompson HS, Corbett JJ . Relative afferent pupillary defects in optic neuritis. Am J Ophthalmol 1981; 92: 685–690.
Kawasaki A . Physiology, assessment, and disorders of the pupil. Curr Opin Ophthalmol 1999; 10: 394–400.
Warner JE, Lessell S, Rizzo III JF, Newman NJ . Does optic disc appearance distinguish ischemic optic neuropathy from optic neuritis? Arch Ophthalmol 1997; 115: 1408–1410.
Goldstein JE, Cogan DG . Exercise and the optic neuropathy of multiple sclerosis. Arch Ophthalmol 1964; 72: 168–170.
Beck RW, Gal RL, Bhatti MT, Brodsky MC, Buckley EG, Chrousos GA et al. Visual function more than 10 years after optic neuritis: experience of the optic neuritis treatment trial. Am J Ophthalmol 2004; 137: 77–83.
Mojon DS, Rösler KM, Oetliker H . A bedside test to determine motion stereopsis using the Pulfrich phenomenon. Ophthalmology 1998; 105: 1337–1344.
Beck RW, Arrington J, Murtagh FR, Cleary PA, Kaufman DI, Optic Neuritis Study Group. Brain MRI in acute optic neuritis: experience of the Optic Neuritis Study Group. Arch Neurol 1993; 8: 841–846.
Optic Neuritis Study Group. The five-year risk of multiple sclerosis after optic neuritis. Experience of the Optic Neuritis Treatment Trial. Neurology 1997; 49: 1404–1413.
The Optic Neuritis Study Group. Multiple sclerosis risk after optic neuritis: final optic neuritis treatment trial follow-up. Arch Neurol 2008; 65: 727–732.
Kupersmith MJ, Alban T, Zeiffer B, Lefton D . Contrast-enhanced MRI in acute optic neuritis: relationship to visual performance. Brain 2002; 125: 812–822.
Rolak LA, Beck RW, Paty DW, Tourtellotte WW, Whitaker JN, Rudick RA . Cerebrospinal fluid in acute optic neuritis: experience of the optic neuritis treatment trial. Neurology 1996; 46: 368–372.
Cole SR, Beck RW, Moke PS, Kaufman DI, Tourtellotte WW . The predictive value of CSF oligoclonal banding for MS 5 years after optic neuritis. Optic Neuritis Study Group. Neurology 1998; 51: 885–887.
Optic Neuritis Treatment Group. Visual function 15 years after optic neuritis. Ophthalmology 2008; 115: 1079–1082.
Goodin DS . Perils and pitfalls in the interpretation of clinical trials: a reflection on the recent experience in multiple sclerosis. Neuroepidemiology 1999; 18: 53–63.
Sellebjerg F, Nielsen HS, Frederiksen JL, Olesen J . A randomized, controlled trial of oral high-dose methylprednisolone in acute optic neuritis. Neurology 1999; 52: 1479–1484.
Chrousos GA, Kattah JC, Beck RW, Cleary PA, The Optic Neuritis Study Group. Side effects of glucocorticoid treatment: experience of the Optic Neuritis Treatment Trial. JAMA 1993; 269: 2110–2112.
McKee MD, Waddell JP, Kudo PA, Schemitsch EH, Richards RR . Osteonecrosis of the femoral head in men following short-course corticosteroid therapy: a report of 15 cases. CMAJ 2001; 164: 205–206.
Roed HG, Langkilde A, Sellebjerg F, Lauritzen M, Bang P, Mørup A et al. A double-blind, randomized trial of IV immunoglobulin treatment in acute optic neuritis. Neurology 2005; 64: 804–810.
Noseworthy JH, O’Brien PC, Petterson TM, Weis J, Stevens L, Peterson WK et al. A randomized trial of intravenous immunoglobulin in inflammatory demyelinating optic neuritis. Neurology 2001; 56: 1514–1522.
Ruprecht K, Klinker E, Dintelmann T, Rieckmann P, Gold R . Plasma exchange for severe optic neuritis: treatment of 10 patients. Neurology 2004; 63: 1081–1083.
Beck RW, Cleary PA, Trobe JD, Kaufman DI, Kupersmith MJ, Paty DW et al. The effect of corticosteroids for acute optic neuritis on the subsequent development of multiple sclerosis. The Optic Neuritis Study Group. N Engl J Med 1993; 329: 1764–1769.
Hickman SJ, Dalton CM, Miller DH, Plant GT . Management of acute optic neuritis. Lancet 2002; 360: 1953–1962.
Hickman SJ, Jay WM, Ko M, Chaudhry F, Plant GT . Optic neuritis: an update typical and atypical optic neuritis. Neuroophthalmology 2008; 32: 237–248.
Chard DT, Dalton CM, Swanton J, Fisniku LK, Miszkiel KA, Thompson AJ et al. MRI only conversion to multiple sclerosis following a clinically isolated syndrome. J Neurol Neurosurg Psychiatry 2011; 82: 176–179.
Swanton J, Fernando K, Dalton C, Miszkiel K, Altmann D, Plant G et al. Early MRI in optic neuritis: the risk for clinically definite multiple sclerosis. Mult Scler 2010; 16: 156–165.
Galetta SL . The controlled high risk Avonex multiple sclerosis trial (CHAMPS Study). J Neuroophthalmol 2001; 21: 292–295.
Comi G, Filippi M, Barkhof F, Durelli L, Edan G, Fernández O et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised study. Lancet 2001; 357: 1576–1582.
Kappos L, Freedman MS, Polman CH, Edan G, Hartung HP, Miller DH et al. Effect of early versus delayed interferon beta-1b treatment on disability after a first clinical event suggestive of multiple sclerosis: a 3-year follow-up analysis of the BENEFIT study. Lancet 2007; 370: 389–397.
Optic Neuritis Study Group. High- and low-risk profiles for the development of multiple sclerosis within 10 years after optic neuritis. Arch Ophthalmol 2003; 121: 944–949.
Chard DT, Dalton CM, Swanton J, Fisniku LK, Miszkiel KA, Thompson AJ et al. MRI only conversion to multiple sclerosis following a clinically isolated syndrome. J Neurol Neurosurg Psychiatry 2011; 82: 176–179.
Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG . The clinical course of neuromyelitis optica (Devic's syndrome). Neurology 1999; 53: 1107–1114.
Wingerchuki DM, Weinshenker BG . Neuromyelitis optica: clinical predictors of a relapsing course and survival. Neurology 2003; 60: 848–853.
Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG . Revised diagnostic criteria for neuromyelitis optica. Neurology 2006; 66: 1485–1489.
Polman CH, Reingold SC, Edan G, Filippi M, Hartung HP, Kappos L et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the ‘McDonald Criteria’. Ann Neurol 2005; 58: 840–846.
Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM et al. A role for humoral mechanisms in the pathogenesis of Devic's neuromyeltis optica. Brain 2002; 125: 1450–1461.
Petzold A, Pittock S, Lennon V, Maggiore C, Weinshenker BG, Plant GT . Neuromyelitis optica-IgG (aquaporin-4) autoantibodies in immune mediated optic neuritis. J Neurol Neurosurg Psychiatry 2010; 81: 109–111.
Pittock SJ, Weinshenker BG, Lucchinetti CF, Wingerchuk DM, Corboy JR, Lennon VA . Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol 2006; 63: 964–968.
Matsushita T, Isobe N, Matsuoka T, Shi N, Kawano Y, Wu XM et al. Aquaporin-4 autoimmune syndrome and anti-aquaporin-4 antibody-negative opticospinal multiple sclerosis in Japanese. Mult Scler 2009; 15: 834–847.
Weinshenker BG, O’Brien PC, Petterson TM, Noseworthy JH, Lucchinetti CF, Dodick DW et al. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann Neurol 1999; 46: 878–886.
Cree B, Lamb S, Chin A, Bonovich D, Islar J, Waubant E et al. Tolerability and effects of rituximab (anti-CD20 antibody) in neuromyelitis optical (NMO) and rapidly worsening multiple sclerosis (MS). Neurology 2004; 62: A492.
Saiz A, Zuliani L, Blanco Y, Tavolato B, Giometto B, Graus F et al. Revised diagnostic criteria for neuromyelitis optica (NMO). Application in a series of suspected patients. Neurol 2007; 254: 1233–1237.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Pau, D., Al Zubidi, N., Yalamanchili, S. et al. Optic neuritis. Eye 25, 833–842 (2011). https://doi.org/10.1038/eye.2011.81
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2011.81
Keywords
This article is cited by
-
Next-generation nanomaterials: advancing ocular anti-inflammatory drug therapy
Journal of Nanobiotechnology (2023)
-
Pregnancy prevalence and outcomes after optic neuritis in South Korea
Scientific Reports (2023)
-
Sub internal limiting membrane hemorrhage followed by bilateral optic disc hemorrhage in Kikuchi-Fujimoto disease: a case report
BMC Ophthalmology (2021)
-
B-cell related biomarkers associated with severity of the first demyelinating event of acute optic neuritis
Eye (2020)
-
Critical Role of Monocyte Recruitment in Optic Nerve Damage Induced by Experimental Optic Neuritis
Molecular Neurobiology (2019)
