
Abstract
Aims
Coats disease is an uncommon form of retinal telangiectasis. Published case series mostly originate from tertiary referrals centres and may provide a skewed view of disease severity. We conducted a prospective population-based study of Coats disease in the United Kingdom to ascertain the incidence and provide a more representative picture.
Methods
The study was conducted through the British Ophthalmological Surveillance Unit. This first paper reports the features at presentation; gender, mode of presentation, visual acuity, anterior and posterior segment findings, amount of retinal exudation, and disease staging.
Results
A total of 55 eligible cases of Coats disease were identified giving an estimated population incidence of 0.09 per 100 000 of the population. All cases were unilateral and 85% were male. Mean age at presentation was 146 months (median 96 months). The mean age of diagnosis was markedly different with differing mechanisms of presentation. Cases presenting with leucocoria or strabismus presented early whereas subjective visual loss presented much later. A large proportion of eyes (44%) were blind at diagnosis. The great majority of eyes (71%) had 6 or fewer clock hours of retinal exudation. More severe forms/stages of Coats disease were more common in the youngest patients.
Conclusions
Compared with published studies of Coats disease, we have found milder disease severity at presentation. This is most likely because of the population-based nature of our study reflecting the full disease spectrum. A large proportion of eyes with Coats have poor visual acuity and disease severity is worse in younger patients.
Similar content being viewed by others


Introduction
Coats disease is a well described but uncommon form of idiopathic retinal telangiectasis that is usually unilateral and affects males more than females.1 The telangiectatic vessels in Coats disease lead to intra-retinal and sub-retinal exudation, in the absence of vitreo-retinal traction.1, 2 This condition was originally described by Coats in 1908, though his series also included eyes with retinal capillary haemangiomas (Von-Hippel Lindau disease).3 In 1956, Reese4 refined the definition and description into the condition we would now recognise as Coats disease.
Although many eyes with Coats can be stabilised, more advanced cases may progress to complete retinal detachment, neovascular glaucoma, and frequently require enucleation.1, 5, 6, 7 While there are a number of published case series of Coats disease, these mostly originate from large tertiary referrals centres and perhaps give a skewed view of the severity of Coats disease.1, 5, 6 We conducted a prospective study of Coats disease in the United Kingdom, which would provide a more representative picture of the full clinical spectrum of Coats disease, current treatment preferences and their anatomic and visual outcomes. This information will hopefully be very informative to clinicians and should allow them to plan treatment for patients with Coats and to counsel patients and parents.
Materials and methods
New cases of Coats disease were prospectively ascertained through the monthly active surveillance system of the British Ophthalmological Surveillance Unit (BOSU).8 At the end of each month the yellow BOSU report card was sent to all consultant and associate specialist ophthalmologists in the United Kingdom (this list is maintained by the BOSU who monitor new appointments and conduct an annual telephone census). The card contains up to 10 rare eye conditions of interest and respondents are asked to indicate all new cases of these conditions (in this case Coats disease), or to confirm that they had no new cases to report. All ophthalmologists reporting new cases of Coats disease to the investigators were sent a baseline questionnaire and a follow-up questionnaire 6 months later. Ethical approval for this study was obtained from Fife, Forth Valley, and Tayside Research Ethics Service, REC reference 06/S0501/43. The ethical approval permitted the investigators to send reminders for unreturned questionnaires though no other contact was permitted.
The Coats disease study was on the BOSU reporting cards for twelve months from January 2008, the one-hundredth anniversary of George Coats original publication.3 The baseline questionnaire enquired about age at presentation, gender, mode of presentation, initial LogMAR visual acuity, and clinical features. The clinical features included anterior and posterior segment findings, number of clock hours of retinal exudation, and disease staging. The staging of Coats disease followed that described by Shields.6 Stage 1 has telangiectasia only; stage 2 is characterised by telangiectasia plus exudation (2A extrafoveal exudation; 2B foveal exudation); stage 3 is characterised by exudative retinal detachment (3A subtotal; 3B total). Stage 4 has total retinal detachment with secondary glaucoma, and stage 5 is advanced end stage disease.6
We enquired about investigations carried out and the initial treatments employed. Finally, we enquired whether there was a family history of eye disease. Patients with associated ophthalmic disorders such as retinitis pigmentosa and underlying medical conditions and syndromes were excluded, as suggested by Sheilds.1, 5
The follow-up questionnaire 6-months later enquired about LogMAR visual acuity, clinical features, disease stage, and whether any additional investigations or treatments were required. Given the volume of information we have reported the data in two separate papers. This paper reports on the epidemiology and clinical features at presentation. The second paper reports on the investigations and treatments performed and their outcomes.
Results
A total of 55 completed baseline questionnaires (from 72 BOSU notifications) for eligible cases of Coats disease were returned to the authors. One additional case of unilateral Coats type retinopathy was excluded because of the underlying Cornelia de Lange syndrome, giving a response rate of 77.8%. The mid 2008 population of the United Kingdom was 61.4 million (http://www.statistics.gov.uk/cci/nugget.asp?id=6), providing a minimum population incidence of 0.09 per 100 000 of the population.
In all, 42 completed 6-month follow-up questionnaires were also returned. All 55 cases were unilateral and comprised 30 right eyes (55%) and 25 left eyes (45%). In all, 27 of the patients were male (85%) and 8 (15%) were female.
Age at diagnosis
The mean age at presentation was 146 months (range 23–888 months) though this figure is skewed by a small number of cases presenting in later life. The median age at presentation of 96 months is more representative as can be seen in Figure 1, which shows the number of cases presenting, clumped together into 5-year blocks. From Figure 1 it can be clearly seen that the great majority of cases present in the first 15 years of life, peaking in the 5–10 years of age group.

Age at detection of Coats.
Reason for referral
Some cases of Coats disease presented because of leucocoria (n=7), strabismus (n=10), or subjectively reported visual loss (n=18). Other cases, however, were detected as a result of pre-school vision screening (n=9), as an incidental finding during an ophthalmic examination (n=3), or as part of an optometric examination (n=8). The mean age of diagnosis was markedly different with the different mechanisms, Figure 2. Cases presenting with leucocoria or strabismus had mean ages of 46 and 52 months, respectively. Cases presenting with subjective visual loss on the other hand had a mean age at presentation of 261 months. In cases presenting with strabismus, the strabismus was presumably secondary to poor vision as all had LogMAR visual acuity ≤1.0 (6/60). The mean age at detection by routine ophthalmic examination, screening, or optometric examinations were, 92 months, 100 months, and 166 months, respectively.

Mean age for different modes of diagnosis of Coats.
Clinical features at diagnosis:
-
a)
Visual acuity: Figure 3 shows the range of visual acuities of cases of Coats disease at diagnosis. A large proportion of eyes (44%) had very poor vision at diagnosis with LogMAR visual acuity of 1.0 (6/60) or worse. Only 19 eyes (35%) had LogMAR visual acuity of 0.3 (6/12) or better.
Figure 3 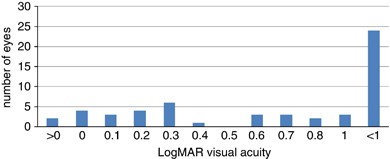
Visual acuity at diagnosis.
-
b)
Anterior segment findings: Only a minority of eyes (n=11; 20%) had abnormal anterior segment findings at baseline examination. Leucocoria was noted in all 11 eyes, including the 7 who had leucocoria as the presenting feature. Additionally, rubeosis was found in one eye and rubeosis plus heterochromia in one additional case.
-
c)
Posterior segment findings: The retinal features of all 55 eyes were classified according to the system proposed by Shields.5 The findings are summarised in Figure 4. It can be seen that there is a very marked clustering in the middle grades of severity (stages 2 and 3) with no eyes in stages 1 or 5 and only four eyes in stage 4.
Figure 4 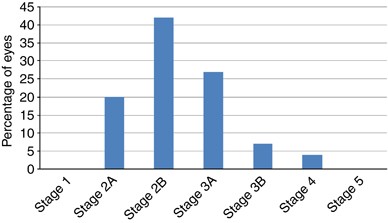
Coats disease stage at diagnosis.
-
d)
Extent of retinal exudation: The great majority of eyes (n=32; 71%) had 6 or fewer hours of retinal exudation at presentation.
-
e)
Is Coats disease worse in younger patients? We plotted those patients with more marked forms of Coats disease according to age at presentation, Figure 5. Given the very small number of eyes with stage 4 or 5 disease (n=2), we looked at all eyes with stage 3 (retinal detachment) or worse at presentation. It can be clearly seen that the stage 3 disease or worse is more common at diagnosis in the youngest patients and becomes less common in older patients.
Figure 5 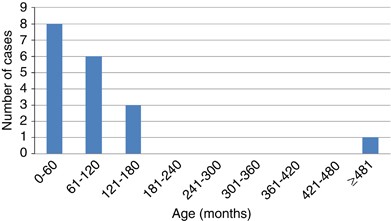
Coats disease stage 3 or worse at diagnosis by age group.
-
f)
Family history of ocular disease: No family history of significant ocular disease was reported for any of the notified cases.



Discussion
Coats disease is an uncommon but well described form of retinal telangiectasis known to most ophthalmologists and has been the subject of several small and large case series.1, 7, 9, 10, 11 While previous studies have been enlightening they did not seem to reflect the cases of Coats disease we have typically encountered in clinical practise. On the other hand, the smaller numbers in our study could make the differences observed to be a chance rather than a real occurrence.
This study confirms that Coats disease is rare, however, surveillance using a single source to identify cases is vulnerable to under ascertainment.8 The incidence of 0.09% per 100 000 is reported as a minimum estimate of incidence, as is the case with other surveillance studies.12 During the study period the BOSU achieved consistently high monthly card return rates (mean 77%) and this along with the 78% study response rate here indicate good compliance with the study.8 The reasons for the response rate of 78% in our study are probably similar to those for all BOSU studies.8 The principal reasons are likely to have been failing to keep a record of the patient's identity and not being able to get hold of the case notes.8
The BOSU employs a prospective active case ascertainment system, which is the best approach to identifying cases of interest. Evaluation trials comparing passive and active surveillance systems consistently report that participants in an active scheme notify around twice as many cases per head of population.13 Furthermore, the serious and complex natures of the diseases under surveillance necessitate the patient to seek the care of a senior ophthalmologist. The inclusion of all ophthalmologists with clinical autonomy in the United Kingdom, therefore, provides a theoretical basis for complete population coverage allowing for meaningful epidemiological analysis.
Consequently, there are good reasons to have confidence in the completeness of reporting and the representativeness of these data.
The most notable series of patients with Coats disease is that reported by Shields et al, with over 150 patients.1, 5, 6 While our series is smaller it is significantly different in several respects and because it is population based, more likely to be representative of the typical spectrum of Coats. Shields reported a mean age at presentation of 11 years (132 months), comparable to our finding of 146 months.1 While our median age at presentation (96 months) is also lower than the mean presentation, it differs significantly from Shields figure of 5 years (60 months). This difference may be because many younger children were referred to the ocular oncology at Wills Eye Hospital because of concern about possible underlying retinoblastoma by the referring doctor (27% of cases).1 Indeed, in a different series of 10 children with Coats disease reported by Char11, and who were originally referred because of suspected retinoblastoma, the mean age at diagnosis was just 2.4 years (29 months).
The data on the mechanism of presentation or diagnosis in our study are both interesting and intuitive. The mean age of diagnosis was markedly different with the different mechanisms, Figure 2. Younger children are most likely to present with leucocoria or strabismus, whereas older children and adults present with subjectively reduced visual acuity or because of detection while the condition is asymptomatic. We believe that these differing mechanisms of presentation with age have not been described previously. Screening was responsible for 16% of diagnoses and a similar proportion were diagnosed incidentally during routine optometric examinations. The mean age at detection by screening was 100 months of age, which doesn’t correspond to the typical age at which vision screening occurs in the United Kingdom (typically 48–60 months of age). One possible explanation is that some cases diagnosed during routine eye examinations were mislabelled as ‘screening’ by the returning doctor.
Coats disease is associated with very poor vision (LogMAR vision ≤1.0) at presentation in a large proportion of cases in all published case series, though the proportion varies. In Shields series, 76% of eyes had very poor vision.1 Cahill et al reported 50% when patients with associated ophthalmic or medical conditions are excluded.7 In the series reported by Budning et al, the comparable figure is 88%.9 Our figure of just 44% of eyes with very poor vision at presentation is the lowest reported. The levels of very poor vision at presentation also concur with the number of clock hours of retinal exudation. In our series, the percentage of eyes with 6 clock hours or less of retinal exudation at diagnosis was 71%. The comparable figures for the series reported by Shields, Cahill, and Budning are 27, 74, and 44%, respectively.1, 7, 9
The anterior segment findings at baseline examination (not just as the presenting feature) in our series reflect the less severe or advanced nature of Coats disease compared with Shields. We found an incidence of leucocoria of 20% while Shields reported 24%.1 Iris neovascularisation was present in just two eyes (4%) in our series and 8% in the Shields series.
Shields et al6 have described a staging classification system for Coats disease based on their extensive experience of 150 patients (158 eyes). Eyes having stage 1 at initial assessment are very rare, just 1% in Shields series6 and none at all in our population-based study. Severe end stage eyes (stage 4 and 5) were rare in our series with just four cases (7%) compared with 17% for Shields.6
It has been reported previously that Coats disease is worse in younger patients. Shields et al found that the extent of retinal detachment was worse in children under three years of age.1 Furthermore, Cahill et al reported a mean age at diagnosis, which was lowest in the group with the most severe disease.7 Figure 5 shows the number of cases in our series that had stage 3 (retinal detachment) or worse at presentation. It can be clearly seen that the more severe stages of Coats disease predominate in the lower age groups. What we cannot say, however, is how much of this increased severity is due to delayed presentation in patients too young to report subjectively reduced visual acuity. Conversely, Coats disease presenting in adulthood is more likely to have less severe disease in terms of staging.10 In a series of Coats disease in adult patients only reported by Smithen et al,10 the exudation was generally posterior to the equator only and involved 6 clock hours or fewer in 77% of cases.
One question that we were unable to answer was whether or not Coats disease can be inherited as there was not a single reported instance of a family history of significant eye disease in our series. Black et al14 have reported the case of a female with unilateral Coats disease who gave birth to a son with Norrie's disease. Both mother and son carried a missense mutation in the NDP (Norrie) gene on chromosome Xp11.2 leading to speculation that mutations in this gene may be responsible for at least some cases of Coats disease.14, 15
We have reported the clinical features at presentation of the spectrum of Coats disease presenting over a 12-month period in the United Kingdom. We have confirmed the findings of previous studies that Coats predominantly affects males and that it is usually unilateral and that it preferentially affects younger patients. Overall, the most notable finding of this study is the milder disease severity compared with existing studies. This is most likely because of the population-based nature of this study and the skewed referral patterns in other series from large tertiary referral centres. It is also possible that earlier diagnosis as a result of parents bringing their children to optometrists for routine eye examinations may have had an effect. The companion paper to this, describes the investigations and treatments employed for Coats disease as well as the anatomic and visual outcomes.

References
Shields JA, Shields CL, Honavar SG, Demirci H . Clinical variations and complications of Coats disease in 150 cases: the 2000 Sanford Gifford Memorial Lecture. Am J Ophthalmol 2001; 131 (5): 561–571.
Chang M, McLean IW, Merritt JC . Coat's disease: a study of 62 histologically confirmed cases. J Pediatr Ophthalmol Strabismus 1984; 21: 163–168.
Coats G . Forms of retinal diseases with massive exudation. Roy Lond Ophthalmol Hosp Rep 1908; 17: 440–525.
Reese AB . Telangiectasis of the retina and Coats disease. Am J Ophthalmol 1956; 42: 1–8.
Shields JA, Shields CL . Coats disease: the 2001 LuEsther T Mertz Lecture. Retina 2002; 22 (1): 80–91.
Shields JA, Shields CL, Honavar SG, Demirci H, Cater J . Classification and management of Coats disease: the 2000 Proctor Lecture. Am J Ophthalmol 2001; 131 (5): 572–583.
Cahill M, O’Keefe M, Acheson R, Mulvihill A, Wallace D, Mooney D . Classification of the spectrum of Coats’ disease as subtypes of idiopathic retinal telangiectasis with exudation. Acta Ophthalmol Scand 2001; 79: 596–602.
Foot BG, Stanford MR, Rahi J, Thompson JR . The British Ophthalmological Surveillance Unit: an evaluation of the first 3 years. Eye 2003; 17: 9–15.
Budning AS, Heon E, Gallie BL . Visual prognosis of Coats disease. J Am Assoc Pediatr Ophthalmol Strabismus 1998; 2: 356–359.
Smithen LM, Brown GC, Brucker AJ, Yannuzzi LA, Klais CM, Spaide RF . Coats disease in adulthood. Ophthalmol 2005; 112 (6): 1072–1078.
Char DH . Coats’ syndrome: long term follow-up. Br J Ophthalmol 2000; 84 (1): 37–39.
Ling R, Cole M, James C, Kamalarajah S, Foot B, Shaw S . Suprachoroidal haemorrhage complicating cataract surgery in the UK: epidemiology, clinical features, management, and outcomes. Br J Ophthalmol 2004; 88: 478–480.
Thacker SB, Redmond S, Berkelman RL . A controlled trial of disease surveillance strategies. Am J Prev Med 1986; 2: 345–350.
Black GC, Perveen R, Bonshek R, Cahill M, Clayton-Smith J, Lloyd IC et al. Coats disease of the retina caused by somatic mutation in the NDP gene: a role for norrin in retinal angiogenesis. Hum Mol Genet 1999; 8: 2031–2035.
Luhmann JF, Lin J, Acar N, Lammel S, Feil S, Grimm C et al. Role of the Norrie disease pseudoglioma gene in sprouting angiogenesis during development of the retinal vasculature. Invest Ophthalmol Vis Sci 2005; 46 (9): 3372–3382.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
This project received funding from the following charitable/non-commercial sources, which exercised no influence over the design of the study or analysis of data: Eyecare fund, Princess Alexandra Eye Pavilion, Edinburgh and Sick Kids Friends Foundation, Royal Hospital for Sick Children, Edinburgh.
Rights and permissions
About this article
Cite this article
Morris, B., Foot, B. & Mulvihill, A. A population-based study of Coats disease in the United Kingdom I: epidemiology and clinical features at diagnosis. Eye 24, 1797–1801 (2010). https://doi.org/10.1038/eye.2010.126
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2010.126
Keywords
This article is cited by
-
Coats disease in India: clinical presentation and outcome in 675 patients (690 Eyes)
International Ophthalmology (2022)
-
Clinicopathological Correlations in Enucleated Globes of Late-Stage Coats Disease with a Review of the Literature
Journal of Epidemiology and Global Health (2022)
-
Clinical features and prognostic factors in 71 eyes over 20 years from patients with Coats’ disease in Korea
Scientific Reports (2021)
-
Recent advances in the diagnosis and treatment of Coats’ disease
International Ophthalmology (2019)
