
Abstract
Aim
To present the clinical features and management outcomes in a large longitudinal series of patients with craniofacial fibrous dysplasia (CFD).
Methods
Retrospective interventional consecutive case series. Main outcome measures included signs and symptoms, radiographic findings, long-term outcomes, and postoperative complications.
Results
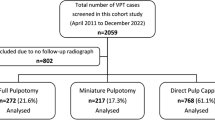
A total of 42 patients with CFD were identified. The mean age at presentation was 16.7 years; mean follow-up was 12.6 years. Out of these 42 patients, 37 (88.1%) had unilateral involvement and 5 (11.9%) had bilateral involvement, of which 3 (7.1%) had McCune–Albright syndrome. The commonest presenting symptom was facial asymmetry (36 cases, 86%). The frontal bone was the most commonly involved (27 cases, 64.3%), zfollowed by the sphenoid (24 cases, 57.1%). The most common pattern of bone involvement was monostotic (32 cases, 76.2%). Radiological optic canal involvement occurred in 18 eyes of 15 (37.5%) patients, with optic atrophy in 9 eyes (18.8%) of 7 patients (16.7%). Surgical intervention was performed in 30 (71.4%) cases for both functional and reconstructive reasons. Optic canal decompression was performed in three cases, in all of which stabilization of vision was achieved; no patient lost vision as a result of surgery.
Conclusions
In this large longitudinal series of CFD, visual loss was not uncommon and occurred insidiously. The presenting clinical and radiological features, surgical interventions, and outcomes are discussed.
Similar content being viewed by others


Introduction
The first description of fibrous dysplasia is attributed to Von Recklinghausen in 1891,1 although it was Lichtenstein who first coined the term ‘fibrous dysplasia’.2 It is an uncommon, histopathologically benign disease, characterized by the replacement of normal marrow by proliferating fibro-osseous tissue, which expands and thins overlying cortex.3 Its aetiology is a post-zygotic, somatic mutation of the GNAS gene, which encodes the α subunit of the ubiquitous stimulatory G protein.4, 5 Fibrous dysplasia is a non-familial disease with onset usually occurring by 10 years of age, although progression is known to occur into adulthood with associated cosmetic and functional morbidity.
Fibrous dysplasia occurs in a polyostotic form in 30% of cases or, more commonly, a monostotic form (70%).6 When limited to the craniofacial region, most authors consider the disease process to be monostotic even if several different bones are affected, as there is only one disease focus.7 Craniofacial involvement occurs in 50% of patients with polyostotic and 27% of patients with monostotic fibrous dysplasia.8 The McCune–Albright syndrome is a subtype of the polyostotic form associated with areas of cutaneous pigmentation and precocious puberty, first described by Albright and associates in 1937.3
Previous reports in the literature on ophthalmic manifestations include case series and numerous descriptive, often cross-sectional studies focusing on the question of prophylactic vs therapeutic optic nerve decompression.9, 10 We present an analysis of a longitudinal series of 42 cases of craniofacial fibrous dysplasia (CFD), comparing their clinical features, radiological patterns, and long-term outcomes, with a mean follow-up period of 12.6 years.
Materials and methods
A retrospective descriptive analysis of the clinical presentation, management, and postoperative course of 42 consecutive patients with radiologically confirmed fibrous dysplasia was conducted. Cases were identified from the Australian Craniofacial Unit database. These patients were managed at the Women's and Children's and the Royal Adelaide Hospitals in South Australia between 1975 and 2006. Ethical approval was obtained from the local institutional review board.
The diagnosis of CFD was established after clinical review by the craniofacial surgeons; all patients had radiological evaluation. A combined team consisting of an ophthalmologist and a craniofacial surgeon evaluated all patients. Histological confirmation of fibrous dysplasia was performed only if operative intervention occurred.
Data from ophthalmology reviews were obtained from the relevant case notes and were documented on standardized pro forma sheets. Exclusion criteria were: (1) previous craniofacial, cranial, or strabismus surgery; and (2) lack of preoperative ophthalmic assessment. The types of data collected are shown in Table 1.
Visual acuity was measured by standardized methods appropriate to age, with patients being considered to have impaired vision (in this study) if they had a visual acuity of 6/12 or less, or more than two Snellen lines poorer than age-adjusted normative values in at least one eye.11 Visual fields were assessed by the Humphrey Visual Field/Swedish Interactive Thresholding Algorithm 30-2 computerized testing (Allergan-Humphrey), or Goldmann perimetry testing. Optic nerve dysfunction was defined as the presence of a characteristic scotoma (or field deficit), or an abnormal result on two of four other tests: visual acuity worse than 6/12, correct identification of fewer than 10 of the 14 Ishihara colour plates, the presence of a relative afferent pupillary defect, or evidence of optic nerve head pallor on fundal examination.
Axial and coronal computerized tomographic (CT) sections were obtained. Where possible, high-resolution images of the orbit were performed using a 0.5 mm helical run from the upper teeth to the top of the frontal sinuses in a 200 mm field of view. Three-dimensional reconstruction was performed on the optic canals that were suspected to be narrow on initial CT scanning and sections perpendicular to the longitudinal axis of the optic canal were generated. The dimensions of the optic waist were measured with digital callipers on the reformatted images and the area of the optic canal was calculated using the method described by Lee et al12 (half the height in mm × half the width in mm × π, where π=3.14).
Surgery was undertaken for functional or reconstructive reasons largely based on criteria outlined in the surgical classification described by Chen and Noordhof13 (Table 2).
All patients were followed up by clinical examination, plain radiographs, and CT scanning, ranging from 2 months to 31 years post-diagnosis (mean 12.6 years). The frequency of follow-up was tailored according to disease aggressiveness, the presence of optic nerve compromise, and the type of intervention performed.
Results
Demographics
A total of 42 patients were identified. The mean age at presentation was 16.7 years (median 15 years; range 0–59 years). There were 20 women (47.6 %) and 22 men (52.4%). Of 42 patients, 36 (85.7%) were Caucasian and 6 (14.3%) were Asian. The mean duration from the first signs or symptoms to the diagnosis of fibrous dysplasia was 5.6 years (median 5 years; range 0–40 years). Thirteen (31.0%) patients were diagnosed after the age of 18 years.
Thirty-seven (88.1%) cases had unilateral craniofacial involvement and five (11.9%) had bilateral involvement, of which three (7.1%) presented with the McCune–Albright syndrome.
Clinical presentation
Data on the presenting signs and symptoms of patients are summarized in Table 1. The most common presentation was facial asymmetry, which occurred in 36 (86%) patients, followed by an orbital or facial mass 25 (60%). The most common ocular presentation was blurred vision, which occurred in 10 (24%) patients, followed by eyelid position abnormalities that occurred in 4 (10%) patients.
Vision
Visual acuity was initially worse than 6/12 in 9 (18.8%) of the 48 examined eyes, due to compressive optic neuropathy in 6 (12.5%) eyes and amblyopia in 3 (6.3%). Three further eyes were diagnosed with optic neuropathy (total nine eyes in seven patients), in two of whom the visual acuity had declined to less than 6/12 by the end of follow-up; in the third patient, visual acuity remained stable. In those eyes with optic neuropathy, the median final visual acuity was counting fingers (range 6/9 to perception of light). In this series, no patient presented with acute loss of vision, with visual loss developing insidiously in all cases. Surgical optic canal decompressions were performed in three cases, one of which was prophylactic and two of which were performed for clinical signs of optic nerve compromise; in these patients, the visual acuity was successfully prevented from declining postoperatively, with follow-up of 3–23 years (mean 11.2 years).
Globe displacement
Proptosis was present in 20 (42.6%) eyes; the mean axial displacement was 2.9 mm (median 2.5 mm, range 1–8 mm). Globe displacement occurred in 34 (72.3%) eyes and the mean non-axial displacement (ie vertical or horizontal dystopia of the globe) was 2 mm (median 2 mm, range 1.5–10 mm).
Radiological findings
The type of radiological bone involvement on the CT scans included: sclerotic in 21 (50%) patients, pagetoid in 11 (26.2%) and cystic in 2 (4.8%) (no mention in archived radiological report, 8 cases).
The most common reported pattern of bone involvement as determined by CT was monostotic (Figure 1), observed in 32 (76.2%) patients, compared to polyostotic disease, observed in 10 (23.8%) patients (Figure 2). Of those with monostotic disease, involvement of multiple bones across suture lines occurred in 22 (68.8%) cases and single bone involvement occurred in 5 (15.6%) (no mention in archived radiological report, 5 cases). Radiological sinus involvement occurred in 22 (52.4%) patients. The frontal sinus was the commonest to be involved (14 (63.6%) patients), followed by the ethmoid (12 (54.5%)), the sphenoid (6 (27.2%)), and the maxillary sinus (5 (22.7%)). Single sinus involvement was the most common pattern occurring in 13 patients, with multiple sinus involvement in 9.

Contrast-enhanced axial computed tomography (CT) scan (bone windows) showing a monostotic variant of craniofacial fibrous dysplasia, centred on the right ethmoid bone and obliterating the right posterior ethmoid sinus cavities.

Axial CT scan (bone windows) showing a polyostotic variant of craniofacial fibrous dysplasia, associated with the McCune–Albright syndrome. The bones are grossly expanded with thinning of the overlying cortex. Note the presence of both cyst-like areas as well as more homogenous areas with a so-called ‘ground-glass’ appearance.
The orbital roof (frontal bone) was the most common site of orbital involvement, occurring in 27 (64.3%) patients, followed by the lateral orbital wall (sphenoid) in 24 (57.1%), with involvement of the floor (maxilla) and medial wall (ethmoid) occurring in 18 (42.9%) patients each.
Radiological involvement of the optic canal was seen in 15 (35.7%) patients, 3 of which had bilateral disease; of these, optic atrophy developed in 9 eyes (18.8% of all eyes affected, 50% of all cases of radiologically involved canals). Of these 18 optic canals, measurements of the optic canal had been performed for 10 canals (inappropriate CT images for performing measurements in other cases), 5 of which had optic neuropathy and 5 did not. On CT scans showing radiological optic canal involvement, the average optic canal waist height and width at the narrowest point was 3.70 × 3.26 mm (area 9.47 mm2) in the group with no optic neuropathy, compared with 3.52 × 3.20 mm (area 8.84 mm2) in the group with optic neuropathy.
Histological findings
Histological confirmation of the disease was performed in all cases that underwent surgery (30 cases, 71.4%; Figure 3). No cases showed malignant transformation over the period of observation.

The typical histology of fibrous dysplasia is shown here from one of the cases that underwent surgical intervention. Curvilinear bony trabeculae with irregular margins are contained within a cellular fibrous background.
Surgical intervention and outcomes
Operative intervention was performed in 30 (71.4%) cases; the median time to surgical intervention was 1 year from diagnosis. Radical resections of orbital bones in various combinations were performed in 23 cases; bone contouring was performed in 4 cases. Surgical optic canal decompression was performed in three cases, one of which was prophylactic. Of all those patients that had surgery, 11 required more than one procedure over the follow-up period.
Of the 18 radiologically involved optic canals, three underwent optic canal decompression. In the two therapeutic decompressions performed for compressive optic neuropathy (both in monostotic disease), the visual acuity did not decline further postoperatively (stable at 6/18 in one case, 6/36 in another). In the third case, a prophylactic decompression was performed in a 13-year-old girl with polyostotic disease; the preoperative acuity was 6/6 and there was no subsequent deterioration or progression of disease. The mean follow-up period for these three patients was 11.2 years (range 3–23 years). Of the 15 involved canals that were not decompressed, optic neuropathy developed in 7 eyes and of these 7, the visual acuity deteriorated in only 2 eyes (6/9 to 6/18 and 6/9 to hand movements); a relative afferent pupillary defect or optic atrophy developed in the remaining 5 patients. In the two patients in whom visual decline was noted, there were no significant differentiating features in their respective patterns of disease or demographics.
Postoperative complications occurred in 15 (50%) patients, including infection, binocular diplopia, and cranial nerve palsies in 3 (10%) patients each; pain in 2 (13.3%); epistaxis, hypertrophic scar, anaemia, and ectropion in 1 (6.7%) each.
Follow-up
The mean follow-up for the population was 12.6 years (range 2 months to 31 years). Over the period of observation, radiological progression of craniofacial bones other than the optic canal occurred in three cases, but clinical deterioration of compressive optic neuropathy occurred in two cases.
Discussion
Craniofacial fibrous dysplasia is a histologically benign disease13, 14; however, our series at a tertiary referral centre demonstrates that its presentation usually occurs early in life and a degree of visual loss is not an uncommon complication (18.8% at presentation in this series). The majority of patients required surgical intervention during the course of the disease, mainly due to facial asymmetry and orbital mass effect. It should, however, be noted that our experience likely reflects the tertiary referral of more severely affected cases.
Disease presentation
Fibrous dysplasia has traditionally been regarded as a disease of childhood, with symptoms developing most often within the first two decades of life. The dysplastic expansion becomes quiescent after patients reach puberty in 60–80% of cases; however, the end point of tumour growth is unpredictable and enlargement has been observed through the seventh decade.14 In our studied population, the mean age at presentation was 16.7 years. Of the 30 patients that underwent surgical intervention, 19 (63.3%) were treated surgically before the age of 18 years (the period of most rapid dysplastic expansion); however, the remaining 11 (36.7%) patients treated surgically were 18 years and older.
The typical picture of CFD depends on the expansion and compression of pathological bone against adjacent structures.6 In the largest reported series of 66 patients, Becelli et al15 found that in 24 (36.3%), the disease was lacking evident symptomatology. In a further series of 27 cases, Yavuzer et al16 found that painless bony enlargement resulting in skeletal deformity and gradual asymmetry was the most common symptom. In our studied population, 36 (76.6%) patients presented with facial asymmetry, although 19% had headache at presentation, considerably less than 7/10 (70%) of cases noted to have ipsilateral headache or orbital pain by Rootman.17
Radiological pattern of involvement
The radiological pattern of bone involvement in this series revealed a predominance of sclerosis; this has been variable among the different published series.10, 18, 19, 20, 22 Rootman et al18 have found the pattern to be equally distributed among the two groups, sclerotic and pagetoid. Others have found pagetoid to be the most common.10 In our series, most patients had monostotic lesions that involved multiple adjacent bones and crossed suture lines, which is in agreement with that reported in other series.19 However, distinction between monostotic and polyostotic forms can be difficult because of the intimate connection of the individual craniofacial bones.20
In two major series, the frontal bone was involved in 58% of cases and the sphenoid in 35%.8 Other reports have found the maxilla to be the commonest bone involved.10 As in our series, Rootman et al18 found that the frontal bone was the commonest involved bone in the orbital region.
Visual loss and optic nerve decompression
Visual disturbance has been reported to occur in up to 18% of cases due to optic canal involvement,8 a figure similar to the prevalence in our series, in which compressive optic neuropathy occurred in nine (18.8%) eyes. Chronic visual loss due to compressive optic neuropathy was the only pattern of presentation of visual loss in our series; this contrasts with other series that have reported a predominance of acute visual loss in fibrous dysplasia.10 In addition, amblyopia was common in our series, leading to a visual acuity of <6/12 in three eyes (6.3%), a figure considerably higher than the normal 2.9%, according to population data for children between 4 and 10 years.21 This high prevalence of amblyopia is hard to explain but may relate to the high degree of facial deformity in our patients.
Optic canal decompression surgery can be either therapeutic or prophylactic, with some surgeons performing contralateral prophylactic surgery at the time of initial therapeutic decompression.9 Despite the optic nerve's putative lack of plasticity, improvement following therapeutic surgery is well documented.9, 10 Nevertheless, the realistic goal of such surgery is to maintain vision and must be balanced against the risk of postoperative visual loss.
The case for prophylactic decompression of the optic canal revolves around the unpredictability of onset of acute visual loss (often due to the development of an aneurysmal bone cyst or mucocoele) and the short amount of time before it becomes permanent,9 although treatment with corticosteroids and urgent surgical decompression may occasionally reverse acute visual loss.10 However, the function of prophylactic surgical intervention is controversial as resection of dysplastic bone carries a risk of surgically induced visual loss.1 Proponents of prophylactic surgery argue that optic canal involvement by fibrous dysplasia heralds visual dysfunction: Chen et al9 found that of 18 patients with clinical or radiological evidence of optic canal involvement, 12 (67%) had some degree of visual loss, with 6 of these 12 having visual acuities of hand motion or less, 1 of whom was bilaterally affected. A recent study by Lee et al12 challenged these findings, showing that encasement of the optic canal in fibrous dysplasia was not correlated with visual loss in 38 patients. However, this was a cross-sectional study, conducted at a point in time, and can also be criticized for selection bias: patients who had previously undergone therapeutic optic nerve decompression (ie those patients with visual loss) were excluded from the study. In another recent cross-sectional case series, Cruz et al7 also found that radiological evidence of apical involvement in fibrous dysplasia (55.9% of apices were narrowed, defined as circumferential narrowing) does not necessarily cause a clinical optic neuropathy; however, it is not clear whether patients, who might have already sustained visual loss, were included in the study. In summary, it is clear that optic canal narrowing per se does not necessarily cause optic neuropathy; from the available data, however, in the absence of aneurysmal bone cysts or mucocoele formation, it is not yet clear whether prophylactic decompression of the optic nerve in fibrous dysplasia is indicated or not.
The relatively high incidence of chronic visual loss as a result of compressive optic neuropathy in our longitudinal case series would seem to support early intervention. However, the majority of our patients with radiological stenoses did not progress over the observation period and in those seven eyes with radiological optic canal involvement and compressive optic neuropathy, who did not undergo surgery, the visual acuity did not deteriorate in four (over a mean follow-up of 19 years). Although only a randomized clinical trial will provide a definitive answer, our current practice is not to perform prophylactic decompression, but to monitor those with radiological involvement of the optic canal.
Other surgery
Surgical treatments can be broadly categorized into two different techniques: a conservative approach, consisting of the shaping of the dysplastic bone tissue, often repeated over time, avoiding the removal of bone (or postponing it until it becomes indispensable); and a radical approach, consisting of removal of the pathological bone tissue and its reconstruction with an autologous bone graft.22 Surgical interventions were performed on 30 (71.4%) patients in our series, with 11 requiring further surgery over the course of follow-up.
Lesions that are expected to demonstrate aggressive behaviour include those associated with excessive hormonal drive (McCune–Albright syndrome) and those associated with growth hormone-producing tumours.9 Of the three cases of McCune–Albright syndrome in our series, a 6-year-old male child did not require operative intervention over 19 years of follow-up, another 6-year-old male child required a transcranial resection at 2 years after diagnosis, and an infant required three debulking procedures over 7 years of observation. All three cases had a sclerotic radiological appearance on CT, which is consistent with an aggressive nature of disease with an earlier onset.
Malignant transformation
Malignant transformation of fibrous dysplasia occurs infrequently, with reported frequencies ranging from 0.4 to 4%.23 None of our cases demonstrated malignant change over the period of observation.
References
Edelstein C, Goldberg RA, Rubino G . Unilateral blindness after ipsilateral prophylactic transcranial optic canal decompression for fibrous dysplasia. Am J Ophthalmol 1998; 126: 469–471.
Lichtenstein L . Polyostotic fibrous dysplasia. Arch Surg 1938; 36: 874–898.
Gass JD . Orbital and ocular involvement in fibrous dysplasia. South Med J 1965; 58: 324–329.
Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM . Activating mutations of the stimulatory G protein in the McCune–Albright syndrome. N Engl J Med 1991; 325: 1688–1695.
Schwindinger WF, Francomano CA, Levine MA . Identification of a mutation in the gene encoding the alpha subunit of the stimulatory G protein of adenylyl cyclase in McCune–Albright syndrome. Proc Natl Acad Sci USA 1992; 89: 5152–5156.
Giordano F, Serio P, Savasta S, Oliveri G, Genitori L . Craniofacial surgery in fibrous dysplasia. J Pediatr Endocrinol Metab 2006; 19 (Suppl 2): 595–604.
Cruz AAV, Constanzi M, Attie de Castro FA, Carols dos Santos A . Apical involvement with fibrous dysplasia: implications for vision. Ophthal Plast Reconstr Surg 2007; 23 (6): 450–454.
Chen YR, Fairholm D . Fronto-orbito-sphenoidal fibrous dysplasia. Ann Plast Surg 1985; 15 (3): 190–203.
Chen YR, Breidahl A, Chang CN, Kurimoto M, Endo S, Onizuka K et al. Extradural optic nerve decompression for fibrous dysplasia with a favourable visual outcome. Neurol Med Chir (Tokyo) 1996; 36: 102–105.
Katz BJ, Nerad JA . Ophthalmic manifestations of fibrous dysplasia. Ophthalmology 1998; 105: 2207–2215.
Negrel AD, Maul E, Pokharel GP, Zhao J, Ellwein LB . Refractive error study in children: sampling and measurement methods for a multi-country survey. Am J Ophthalmol 2000; 129 (4): 421–426.
Lee JS, Fitzgibbon E, Butman JA, Dufresne CR, Kushner H, Wientroub S et al. Normal vision despite narrowing of the optic canal in fibrous dysplasia. N Engl J Med 2002; 347: 1670–1676.
Chen YR, Noordhoff MS . Treatment of craniomaxillofacial fibrous dysplasia: how early and how extensive? Plast Reconstr Surg 1990; 86 (5): 835–842; discussion 843–4.
Moore AT, Buncic JR, Munro IR . Fibrous dysplasia of the orbit in childhood. Clinical features and management. Ophthalmology 1985; 92 (1): 12–20.
Becelli R, Perugini M, Cerulli G, Carboni A, Renzi G . Surgical treatment of fibrous dysplasia of the cranio-maxillo-facial area. Review of the literature and personal experience form 1984 to 1999. Minerva Stomatol 2002; 51: 293–300.
Yavuzer R, Bone H, Jackson IT . Fronto-orbital fibrous dysplasia. Orbit 2000; 19 (2): 119–128.
Rootman J . Diseases of the Orbit. 2nd ed. Lippincott, Williams & Wilkins: Philadelphia, 2003, (chapter 9).
Rootman J, Kemp E, La pointe JS . Orbital tumors originating in bone. In: Rootman J (ed). Diseases of the Orbit. A Multidisciplinary Approach. JB Lippincott: Philadelphia, 1988, pp 354–379.
DiCaprio M, Enneking W . Fibrous dysplasia. Pathophysiology, evaluation, and treatment. J Bone Joint Surg Am 2005; 87: 1848–1864.
Delap TG, Kaberos A, Dowling P, Rees J, Golding-Wood D . Cranio-facial fibro-osseous dysplasia. A case report and overview of the management of the condition. Int J Pediatr Otorhinolaryngol 1996; 37 (2): 179–191.
Kvarnstrom G, Jakobsson P, Lennerstrand G . Visual screening of Swedish children: an ophthalmological evaluation. Acta Ophthalmol Scand 2001; 79 (3): 240–244.
Ricalde P, Horswell BB . Craniofacial fibrous dysplasia of the fronto-orbital region: a case series and literature review. J Oral Maxillofac Surg 2001; 59 (2): 157–167; discussion 167–8.
Ruggieri P, Sim FH, Bond JR, Unni KK . Malignancies in fibrous dysplasia. Cancer 1994; 73: 1411–1424.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rahman, A., Madge, S., Billing, K. et al. Craniofacial fibrous dysplasia: clinical characteristics and long-term outcomes. Eye 23, 2175–2181 (2009). https://doi.org/10.1038/eye.2009.6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2009.6
Keywords
This article is cited by
-
Fibrous dysplasia with secondary aneurysmal bone cyst—a rare case report and literature review
Oral and Maxillofacial Surgery (2019)
-
Simultaneous decompression of the orbital lateral wall and optic canal for fibrous dysplasia in early adolescence
SpringerPlus (2016)
-
Fibrous dysplasia of the temporal bone secondary to ear surgery: a case report
Journal of Medical Case Reports (2015)
-
An epidemiological and clinical analysis of craniomaxillofacial fibrous dysplasia in a Chinese population
Orphanet Journal of Rare Diseases (2012)
