
Abstract
Purpose
Joubert syndrome (JS) is an autosomal-recessive inherited complex malformation of the midbrain–hindbrain. It has been associated with ocular and oculomotor abnormalities. The aim of our study was to extend the ophthalmic knowledge in JS and to add new findings.
Methods
In a retrospective study, 10 consecutive patients, who met the revised diagnostic criteria of JS were included. Mutation analysis was carried out in all the cases. Each patient underwent a comprehensive neuro-ophthalmological examination.
Results
Bilateral drusen of the optic disc were found in two patients. Four patients showed bilateral morphological and functional signs of retinal dystrophy (CEP290mutation in two cases and AHI1mutation in one case). In nine patients performance during smooth pursuit, saccades, and vestibulo-ocular reflex (VOR) cancellation was poor.
Conclusions
To the best of our knowledge, the association of optic disc drusen with JS has not yet been described. In support of the earlier findings, decreased smooth pursuit and VOR cancellation, as well as partial-to-complete oculomotor apraxia seem to be the key oculomotor features of JS. Genotype–phenotype correlations showed the predictive value of CEP290and AHI1mutations for retinal involvement.
Similar content being viewed by others
Introduction
Joubert syndrome (JS) is a rare autosomal-recessive disorder, which is characterized by midbrain–hindbrain malformations mainly in the form of agenesis or dysgenesis of cerebellar vermis.1, 2 Diagnostic criteria in JS include hypotonia, ataxia, global developmental delay, and the neuroradiological finding of ‘molar tooth sign’.3, 4 The term ‘JS-related disorders’ was introduced referring to a group of pleiotropic conditions presenting the pathognomonic features of JS associated with variable involvement of other organs and systems. Almost all JS-related disorder genes, so far identified, encode for proteins expressed in the primary cilium or in the centrosome. This observation has linked these disorders to the field of ciliopathies.5
Ocular and oculomotor involvement is common in JS.4, 6, 7, 8, 9, 10 However, findings differ considerably because of a wide range of phenotypic variability among patients with JS.
Materials and methods
In a retrospective study, 10 consecutive patients, who met the revised diagnostic criteria of JS were included.3 Patients' (three females) ages ranged from 8 to 29 years (mean: 18.2 years). Each patient underwent a comprehensive neuro-ophthalmological examination including assessment of ocular alignment and motility.
Results
The clinical data for each patient are summarized in Tables 1 and 2.
Ocular findings
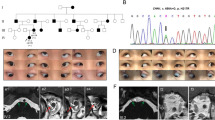
Bilateral drusen of the optic disc were found in patients 5 and 6. Drusen were confirmed by B-scan ultrasound and the autofluorescence phenomenon (Figure 1). In four patients the pigmentation of the peripheral retina was mottled and visual acuity was decreased (Patient 3, 4, 5 and 8).

(a) Colour fundus photograph of the right eye showing optic disc drusen (patient 5). (b) Autofluorescence of the right eye showing optic disc drusen (patient 5).
Oculomotor findings
In nine of the patients, performance during smooth pursuit, saccades, and vestibulo-ocular reflex (VOR) cancellation was poor. The speed in smooth pursuit was uniformly low, catch-up saccades were frequently observed. Abnormal saccades were characterized by increased latency and hypometry if oculomotor apraxia was not complete. In four patients, head thrusts, sometimes with a blink, were used to shift gaze. In four patients, manifest deviations were found. Three of these patients had an alternating esotropia, whereas one had an alternating exotropia.
Discussion
It is surprising that we found bilateral optic disc drusen in two patients. The prevalence of optic nerve drusen has been estimated between <0.4% and 3.7%.11 Both affected patients had signs of retinal dystrophy. The association between drusen and retinitis pigmentosa is well established and the incidence of the combination has been reported to be between 0 and 10%.11 The association between JS and optic disc drusen has not been earlier reported. However, as optic disc drusen are commonly seen in clinical routine, random coincidence cannot be excluded.
Pigmentary fundus changes, retinal dystrophy, and severe visual loss are frequently found in JS.6, 8, 9, 10 Four of our patients had such findings. Genotype–phenotype correlations have emphasized the predictive value of specific mutations for retinal involvement. Retinal involvement is present in about 80% of the patients with AHI1 (patient 8) and CEP290 mutations (patients 3 and 4).5 Genotype–phenotype correlations may even link JS patients with optic disc drusen to specific genes, as MFRP mutations have been identified causing the syndrome of nanophthalmus, retinitis pigmentosa, foveoschisis, and optic disc drusen.12 Other ocular findings such as cataract (patient 10), optic nerve hypoplasia, chorioretinal coloboma, ptosis, and ocular fibrosis are infrequently associated with JS.
Manifest deviations were observed in four out of 10 patients. Ocular misalignment is common in JS.7, 9, 10 Lambert et al.7 described an alternating hyperdeviation on abduction as a characteristic feature, which was seen in two of our patients. In nine of the patients, performance during smooth pursuit, saccades, and VOR cancellation was poor. Our findings of ocular motor defects are in agreement with the findings of earlier studies.4, 7, 9, 10 Defects in smooth pursuit and VOR cancellation can be contributed to deformed cerebellar vermis. Saccades and quick phases of nystagmus are generated involving the brainstem. The control of saccades is regulated by cerebral and cerebellar structures including the frontal eye field and the superior colliculus. Accordingly, lesions of cerebellar control centres lead to dysmetric and slow saccades.
In conclusion, patients were affected by complex, but variable, ocular and oculomotor deficits. This phenotypic variability might be supported by multiplex neuropathological findings of the cerebellar vermis and of several pontine and medullary structures. Furthermore, it may arise from the wide genetic heterogeneity of JS and related disorders, as only about 25% of causative genes and loci have been detected today.
References
Joubert M, Eisenring JJ, Robb JP, Andermann F . Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology 1969; 19: 813–825.
Boltshauser E, Isler W . Joubert syndrome: episodic hyperpnea, abnormal eye movements, retardation and ataxia, associated with dysplasia of the cerebellar vermis. Neuropadiatrie 1977; 8: 57–66.
Maria BL, Boltshauser E, Palmer SC, Tran TX . Clinical features and revised diagnostic criteria in Joubert syndrome. J Child Neurol 1999; 14: 583–590. discussion 590–591.
Maria BL, Hoang KB, Tusa RJ, Mancuso AA, Hamed LM, Quisling RG et al. Joubert syndrome' revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol 1997; 12: 423–430.
Valente EM, Brancati F, Dallapiccola B . Genotypes and phenotypes of Joubert syndrome and related disorders. Eur J Med Genet 2008; 51: 1–23.
Moore AT, Taylor DSI . A syndrome of congenital retinal dystrophy and saccade palsy—a subset of Leber's amaurosis. Br J Ophthalmol 1984; 68: 421–431.
Lambert SR, Kriss A, Gresty M, Benton S, Taylor D . Joubert syndrome. Arch Ophthalmol 1989; 107: 709–713.
Steinlin M, Schmid M, Landau K, Boltshauser E . Follow-up in children with Joubert syndrome. Neuropediatrics 1997; 28: 204–211.
Tusa RJ, Hove MT . Ocular and oculomotor signs in Joubert syndrome. J Child Neurol 1999; 14: 621–627.
Hodgkins PR, Harris CM, Shawkat FS, Thompson DA, Chong K, Timms C et al. Joubert syndrome: long-term follow-up. Dev Med Child Neurol 2004; 46: 694–699.
Davis PL, Jay WM . Optic nerve head drusen. Semin Ophthalmol 2003; 18: 222–242.
Ayala-Ramirez R, Graue-Wiechers F, Robredo V, Amato-Almanza M, Horta-Diez I, Zenteno JC . A new autosomal recessive syndrome consisting of posterior microphthalmus, retinitis pigmentosa, foveoschisis, and optic disc drusen is caused by a MFRP gene mutation. Mol Vis 2006; 12: 1483–1489.
Author information
Authors and Affiliations
Corresponding author
Additional information
Proprietary interest: None
Rights and permissions
About this article
Cite this article
Sturm, V., Leiba, H., Menke, M. et al. Ophthalmological findings in Joubert syndrome. Eye 24, 222–225 (2010). https://doi.org/10.1038/eye.2009.116
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2009.116
Keywords
This article is cited by
-
Macular staphyloma in patients affected by Joubert syndrome with retinal dystrophy: a new finding detected by SD-OCT
Documenta Ophthalmologica (2018)
-
Infantile-Onset Saccade Initiation Delay (Congenital Ocular Motor Apraxia)
Current Neurology and Neuroscience Reports (2015)
-
Visual field defects due to optic disk drusen in children
Graefe's Archive for Clinical and Experimental Ophthalmology (2013)
-
Modelling a ciliopathy: Ahi1 knockdown in model systems reveals an essential role in brain, retinal, and renal development
Cellular and Molecular Life Sciences (2012)
