
Abstract
NHERF1/EBP50 (Na+/H+ exchanger regulating factor 1; Ezrin-binding phosphoprotein of 50 kDa) organizes stable protein complexes beneath the apical membrane of polar epithelial cells. By contrast, in cancer cells without any fixed polarity, NHERF1 often localizes in the cytoplasm. The regulation of cytoplasmic NHERF1 and its role in cancer progression remain unclear. In this study, we found that, upon lysophosphatidic acid (LPA) stimulation, cytoplasmic NHERF1 rapidly translocated to the plasma membrane, and subsequently to cortical protrusion structures, of ovarian cancer cells. This movement depended on direct binding of NHERF1 to C-terminally phosphorylated ERM proteins (cpERMs). Moreover, NHERF1 depletion downregulated cpERMs and further impaired cpERM-dependent remodeling of the cell cortex, suggesting reciprocal regulation between these proteins. The LPA-induced protein complex was highly enriched in migratory pseudopodia, whose formation was impaired by overexpression of NHERF1 truncation mutants. Consistent with this, NHERF1 depletion in various types of cancer cells abolished chemotactic cell migration toward a LPA gradient. Taken together, our findings suggest that the high dynamics of cytosolic NHERF1 provide cancer cells with a means of controlling chemotactic migration. This capacity is likely to be essential for ovarian cancer progression in tumor microenvironments containing LPA.
Similar content being viewed by others


Introduction
The epithelial–mesenchymal transition (EMT) is the process of transdifferentiation of epithelial cells into motile mesenchymal cells.1 During the EMT associated with tumor progression, epithelial cells lose their intrinsic features, including cell-to-cell junctions and apical–basal polarity, and undergo significant morphological changes to increase their potential for dissemination, motility and invasion. This transition is accompanied by significant changes in expression, subcellular localization and functions of multiple protein organizers that define signaling pathways, cytoskeletal structure and lipid composition at the plasma membrane. Many studies have elucidated the key organizers that mediate the diversity, polarity and dynamicity of the cortical structure of cancer cells.
NHERF1 (Na+/H+ Exchanger Regulatory Factor), also known as EBP50 (Ezrin-Binding Phosphoprotein 50) is a member of the NHERF family.2, 3, 4 NHERF proteins are highly expressed in several epithelial tissues and localize at the apical plasma membrane of polar epithelia, which regulates apical microvilli formation.5 An independent study identified NHERF1 as a binding protein of Ezrin/Radixin/Moesin (ERM) proteins,6 which play a crucial role in organizing membrane domains through their ability to interact with transmembrane proteins and the underlying cytoskeleton at the apical membrane.7 Genetic ablation of NHERF1 or ERM results in malformation of apical microvilli, further supporting the idea that the apical protein complex has a significant function in microvillar structure and regulation of polar epithelium.8, 9 Furthermore, the levels of ERM proteins were significantly and specifically lower in the cortical brush border membranes of kidney and small intestine epithelial cells of NHERF1 KO mice. In addition, NHERF1 depletion in cultured cells significantly reduces, but does not eliminate, the microvilli structure on the cell cortex,10, 11 which is reversed by the expression of an NHERF1 construct.12
Like other scaffolding proteins, NHERF proteins contain multiple protein interaction modules, including two N-terminal tandem PDZ domains and a C-terminal ERM-binding domain.6, 13, 14 The PDZ domains interact with a wide variety of transmembrane proteins, including growth factor receptors, G-protein-coupled receptors, ion channels, cell adhesion molecules and cytosolic signaling molecules such as phospholipase C, protein kinases and ERM family proteins.15, 16, 17, 18 NHERF1 serves as a molecular scaffold that mediates the formation of a functional unit by bringing multiple binding partners together into a protein complex. Through these molecular interactions, NHERF proteins play prominent roles as regulators of transmembrane signal transduction, cytoskeletal reorganization, receptor trafficking and phosphoinositide metabolism.18, 19, 20
Lysophosphatidic acid (LPA) is involved in biological responses such as rapid actin rearrangement, stimulation of cell proliferation, suppression of apoptosis and induction of tumor cell invasion.21, 22 LPA induces these responses by binding to members of the LPA receptor (LPAR) family.21, 23 Several lines of evidence have implicated the LPA/LPAR axis in the pathology of human cancers. Since the initial identification of oleoyl-LPA as the crucial molecule in fetal calf serum (FCS) that promotes rat hepatoma cell invasion,24 LPA has also been shown to be the bioactive lipid component of ovarian cancer activating factor (OCAF), which stimulates ovarian cancer cells.25 Notably, the level of LPA is significantly elevated in ascites and sera of most patients with ovarian cancer with poor prognostic outcome.22, 25, 26, 27, 28 In addition, LPA is associated with the EMT: LPARs are highly upregulated during EMT, rendering post-EMT cells more responsive to LPA.29 Furthermore, LPA promotes the migratory and invasive properties of cancer cells,21, 22 eventually resulting in metastasis and poor clinical outcome.22, 30, 31
NHERF proteins are closely associated with tumor development. Initially, NHERF1 was proposed to be a tumor suppressor, in part based on its reduced expression32 and allelic loss33 in various tumor types. However, other studies argued that NHERF1 contributes to cancer progression, based on analyses of large numbers of tumor samples34, 35, 36 and in vitro models using tumor cell lines.37 Despite persistent controversy regarding NHERF1 expression in cancer cells, consensus has emerged regarding the altered subcellular distribution of NHERF1 in the tumor mass and cultured cancer cell lines.32, 38, 39, 40, 41 Specifically, an abnormal transition of NHERF1 localization from the apical membrane of normal epithelium to the cytoplasm, or even the nucleus, has been observed during the EMT in various kinds of tumors. This relocalization is associated with cancer progression, metastatic potential, poor prognosis and estrogen responsiveness, especially in carcinomas. However, despite mounting evidence regarding the pathogenic role of cytosolic NHERF1, little is known about how cytosolic NHERF1 in post-EMT cells behaves in response to the extracellular microenvironment.
In this study, we investigated the regulation of cytosolic NHERF1 in response to LPA stimulation and its contribution to chemotactic cell migration of ovarian cancer cells under a given tumor microenvironment.
Materials and methods
Materials
Chemical reagents
Lysophosphatidic acid (1-oleoyl-2-hydroxy-sn-glycerol-3-phosphate) was purchased from Enzo Life Sciences (Farmingdale, NY, USA). Synthetic siRNA duplexes and Lipofectamine 2000 were from Dharmacon (Lafayette, CO, USA) and Life Technologies (Carsbad, CA, USA), respectively. Cell culture dishes and plates were obtained from BD Bioscience (San Jose, CA, USA). The 96-well-modified Boyden chambers (polycarbonate membranes) were from NeuroProbe (Cabin John, MD, USA); two different pore sizes were used, 8 and 5 μm for the Transwell migration assay and pseudopodia formation assay, respectively. The serum and medium were obtained from Life Technologies, and other chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA) in extra-pure or higher grade.
Antibodies
Rabbit polyclonal α-NHERF1 and α-NHERF2 Ab were generated by immunization of the 6xHis-tagged C-terminal fragment of NHERF1 or −2 (CT: aa 261–351) as described previously.18 α-ERM (catalog no. #3142), α-cpERM (catalog no. #3141), α-pGSKα/β (catalog no. #9331), α-pERK1/2 (catalog no. #3179) and α-pAKT (catalog no. #4060) antibodies were obtained from Cell Signaling Technology (Danvera, MA, USA). α-VSVG antibody was from Sigma-Aldrich.
Plasmid construction
Various plasmid constructs were generated by PCR methods. Full-length cDNAs were PCR amplified using primer pairs for hNHERF1 with appropriate overhang for enzyme cleavage. The PCR products for hNHERF1 WT (aa 1–351), and its deletion fragments (ΔCT: aa 1–260, CT: aa 261–351), were digested with BamHI/XhoI and ligated into pEGFP-C1 or pET30a (+). For cloning into pCMV2-FLAG, PCR products were digested with BglII/XhoI and ligated into pCMV2-FLAG digested with BglII/SalI. GFP-tagged NHERF1-WT was amplified from pEGFP-C1-hNHERF1-WT, and the amplicons were digested with NotI/ClaI and cloned into pAvCMV3 to generate recombinant adenovirus. VSVG epitope-tagged Ezrin WT and its T567A mutant in vector pCB6+ were generous gifts from Dr Monique Arpin. Each cDNA was amplified with NotI/ClaI restriction site overhangs and cloned into pAvCMV3. All mutations were verified by automated sequencing.
Cell culture and transfection
OVCAR-3, SK-OV-3 and MDA-MB-231 cells were from the American Type Culture Collection (ATCC, Manassas, VA, USA). These cell lines were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with penicillin (50 units per ml), streptomycin (50 units per ml) and 10% fetal bovine serum (FBS, Life Technologies) at 37 °C in 95% air/5% CO2. HIO-80, the immortalized human normal ovarian surface epithelial cell line, was a kind gift from Dr Andrew Godwin at the Fox Chase Cancer Center, Philadelphia, PA; these cells were cultured in medium 199 and MCDB 105 (1:1) supplemented with 15% FBS, 0.25 U ml−1 insulin, and 2 mM L-glutamine. For transient transfection, cells were plated into 100 mm dishes and transfected with either siRNA duplexes or plasmid constructs using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
Agonist treatment and cell harvest
Cells were seeded into six-well plates at a density of 1.5 × 106 cells per well. After serum deprivation for 24 h, the cells were treated with LPA as indicated. Each plate was then washed twice with cold phosphate-buffered saline (PBS), and the cells were lysed in 150 μl of cold lysis buffer containing 50 mM HEPES, 150 mM NaCl, 50 mM NaF, 1 mM EDTA/EGTA, 1 mM Na3VO4 and 10% v/v glycerol. Lysates were mildly sonicated and cleared by centrifugation at 4 °C. Protein concentration was determined by BCA assay (Thermo Fisher Scientific, Waltham, MA, USA).
Knockdown by siRNA transfection
SiRNA duplexes targeting NHERF1 (451–469) and NHERF2 (862–880) were synthesized by GE Dharmacon as described in a previous study.18 Pre-synthesized control siRNA duplexes (Luciferase GL3 Duplex) were also purchased and used as controls. Cultured cells were transfected with 20 nM oligonucleotide using Oligofectamine (Thermo Fisher Scientific) under serum-free conditions. Four hours after transfection, cells were washed and supplemented with fresh media containing 10% FBS. Cells were incubated for 72 h before use in experiments.
Recombinant adenovirus production and infection
Recombinant adenovirus expressing GFP-tagged NHERF1 or VSVG-tagged Ezrin (WT or T567A mutant) was generated and amplified in HEK-293T cells and purified by CsCl gradient centrifugation according to the standard protocol.42 OVCAR-3 cells grown to confluence in six-well plates were infected at a multiplicity of infection (MOI) of 10 for 4 h in serum-containing media. In parallel, monolayers were infected with adenovirus containing empty shuttle vector, as a negative control for any nonspecific effect of viral infection on cell viability.
Immunoprecipitation and western blot analysis
Cells were lysed in the lysis buffer and the lysates were allowed to immunoreact with the appropriate antibodies or α-FLAG affinity resin. Immune complexes were collected by centrifugation at 1000 g, and then washed four times with 1 ml of lysis buffer. The resultant precipitates were subjected to SDS-polyacrylamide gel electrophoresis and analyzed by western blot analysis.
Prior to SDS-polyacrylamide gel electrophoresis, all samples were boiled in Laemmli buffer and separated on 6–16% gels according to the standard protocols. Resolved proteins were transferred to nitrocellulose membranes. Membranes were blocked for 1 h in T-TBS buffer containing 10 mM Tris-HCl (pH 7.6), 150 mM NaCl, 0.1% Tween-20 and 5% non-fat milk. Western analyses of OVCAR-3 and MDA-MB-231 cell lysates were probed with antibodies as indicated in each figure. Blots were developed using enhanced chemiluminescence solution (ECL, GE Healthcare; Little Chalfont, Buckinghamshire, UK).
Immunocytochemistry
OVCAR-3 cells were seeded onto cover glasses coated with 10 μg ml−1 collagen type I (Sigma-Aldrich). Cells were transfected with either siRNA duplexes or cDNA constructs, and then serum-deprived for 24 h in DMEM supplemented with 0.1% fatty acid-free BSA. The serum-starved cells were treated with LPA pre-diluted in 0.1% BSA as indicated in figure legends. Cells were washed with PBS and fixed for 20 min at room temperature with 3.7% paraformaldehyde in PBS. Fixed cells were rinsed with TBS (50 mM Tris-HCl (pH 7.4), 150 mM NaCl) and incubated with the permeabilization buffer (PBS supplemented with 0.2% Triton X-100) for 10 min. Cells were rinsed three times for 5 min each with TBS, and then incubated with blocking solution (10% horse serum, 1% BSA, and 0.02% NaN3 in PBS) for 1 h at room temperature. The cells were then stained with α-ERM, α-cpERM and α-VSVG antibodies, and nuclear DNA was stained with Hoechst 33342. Images were captured with simultaneous recording of multiple channels using confocal microscopy (LSM510, Carl Zeiss, Oberkochen, German). Images were digitized using Zeiss imaging software and compiled using Adobe Photoshop Software (Adobe Systems, San Jose, CA, USA).
Cell migration assays using modified Boyden chambers
Cell migration assays were performed as previously described43 with minor modifications. Briefly, cell migration assays were performed using modified Boyden chambers (tissue culture-treated, 5.7 mm diameter, 8 μm pores, ChemoTX; NeuroProbe; Gaithersburg, MD, USA) containing polycarbonate membranes. The underside of the membrane of the upper chamber was coated with 10 μg ml−1 collagen type I (Thermo Fisher Scientific) overnight at 4 °C, rinsed once with 1 × PBS, and then placed into the lower chamber containing 300 μl of migration buffer (serum-free basal medium, with 0.1% fatty acid-free BSA). The cells were removed from culture dishes with diluted trypsin/EDTA (PBS, 0.01% trypsin, and 5 mM EDTA), washed twice with migration buffer, and then resuspended in migration buffer (5 × 105 cells per ml). Next, 2.5 × 104 cells were added to the top of each migration chamber and allowed to migrate to the underside of the top chamber for 3 h. Non-migratory cells on the upper membrane surface were removed with a cotton swab, and the migratory cells attached to the bottom surface of the membrane were fixed in fixation buffer (PBS containing 2% paraformaldehyde), incubated with 0.2% Triton X-100 solution for 10 min at room temperature, and then stained with Hoechst 33342 diluted in PBS. After three washes with PBS, the membrane was excised from the frame and mounted onto the slide glasses. The numbers of migratory cells per membrane were counted with an inverted fluorescence microscope using a × 40 objective. Each determination represents the average of three individual wells, and error bars represent s.d.
Immunostaining of the pseudopodia of the migratory cells
Pseudopodia were generated at the beginning of the migration process, prior to cell body passage through the porous membrane of the ChemoTX chamber. To allow efficient formation of protruding pseudopodia, the cells were detached from the culture dish, resuspended in migration buffer, mounted onto the porous transparent membrane of a ChemoTX chamber (5 μm pore size) at a density of 1 × 104 cells per well, and incubated for 1 h (instead of 3 h as in the standard migration assay). The membrane was washed briefly with ice-cold 1 × PBS containing phosphatase inhibitors. The cells were then immediately fixed with 2% PFA solution and permeabilized for 10 min at room temperature with 0.2% Triton X-100 solution. After soaking in blocking solution for 1 h, the membrane was incubated overnight with α-cpERM antibody diluted in blocking solution. For nuclear staining, the membrane was incubated for 30 min at room temperature with 1 μg ml−1 propidium iodide along with 10 μg ml−1 RNase A. After the final wash, the membrane was excised from the frame, mounted on slide glass, and covered with a cover glass. Using confocal microscopy (LSM510-Meta, Zeiss), images of the cell body and pseudopodia were captured in the same region with different planes of focus, but with constant settings including contrast and gain.
Statistical procedure
All quantitative data are presented as mean ±s.e.m. Two group comparisons were done by two-tailed, unpaired Student’s t-test. Multiple group comparisons were assessed using a one-way or two-way ANOVA, followed by the post hoc Bonferroni test, respectively, when significant main effects or interactions were detected. Statistical significance was set at P<0.05. All comparisons were performed with Prism Software (GraphPad Software, SanDiego, CA, USA).
Results
NHERF1 translocates from cytosol to surface protrusions in response to LPA stimulation
Although NHERF1 has been observed in cortical actin-rich structures such as membrane ruffles, filopodia and microvilli, it remained unknown whether the subcellular localization of NHERF1 is dynamically regulated by extracellular stimuli. In this study, we examined the effect of LPA stimulation on NHERF1 localization in ascites-derived OVCAR-3 ovarian cancer cells. In serum-deprived cells, NHERF1 localized diffusely at the cytosol, whereas LPA stimulation induced rapid NHERF1 translocation to the plasma membrane at the cell margin (Figure 1a). Interestingly, over time following LPA stimulation, OVCAR-3 cells underwent marked morphological changes, generating protruding structures on the cell surface. These protrusions were distributed throughout the plasma membrane of LPA-exposed cells (Figure 1b). NHERF1, which translocated from cytosol to the cell margin, was targeted to these structures. This observation highlights the dynamic relocalization of cytosolic NHERF1 in cancer cells without fixed polarity, in stark contrast to its static apical localization in polar epithelial cells.8

NHERF1 translocates from cytosol to plasma membrane upon LPA stimulation, and then to surface protrusions. (a) Time-lapse imaging of LPA-induced NHERF1 translocation. OVCAR-3 cells were plated onto type I-coated glass-bottom dishes and transfected with GFP-tagged NHERF1 construct. After serum deprivation for 24 h, the cells were incubated in Phenol Red-free DMEM and stabilized for 20 min. After adjusting the plane of focus near the bottom region close to the glass surface, time-lapse scanning was performed at 2 min intervals after 1 μM LPA stimulation. Scale bar, 20 μm. Relative intensities of GFP fluorescence are shown along the broken line of the long arrow (yellow) overlaid on each image, using the line scan tool of Image J software. The relative distribution of GFP-NHERF1 at each time point is plotted in the graph in the right lower corner. (b) High-resolution image of NHERF1 translocation from cytosol to surface protrusions in response to LPA stimulation. OVCAR-3 cells expressing GFP-NHERF1 were stimulated with 1 μM LPA, fixed, and observed by confocal microscopy. The focuses of the confocal imaging are adjusted to the top and the bottom region sequentially, as indicated. Scale bar,20 μm. Relative intensities of GFP fluorescence intensities are shown along the broken line of the long arrow (yellow) overlaid on each image. The relative distribution of GFP-NHERF1 is plotted in the graph immediately below each image.
LPA-induced NHERF1 translocation is mediated by direct interaction with cpERM proteins at the cell margin
Based on the established interaction between NHERF1 and active ERM proteins at the apical microvilli of polar epithelia,5, 8, 19, 44 we postulated that NHERF1 translocation in cancer cells might depend on a conserved interaction with active ERM proteins. Interestingly, LPA stimulation has been shown to induce ERM phosphorylation in Swiss 3T3 fibroblast cells.45 To test this idea, we first investigated whether ERM proteins in OVCAR-3 cells are phosphorylated by LPA stimulation, an established marker of ERM activation.46 LPA stimulation of OVCAR-3 cells resulted in rapid ERM phosphorylation at the conserved threonine residue in a dose-dependent manner (Figure 2a). In addition, co-immunoprecipitation of LPA-stimulated OVCAR-3 cell lysates revealed that NHERF1 interacts with cpERM proteins (Figure 2b), confirming the direct interaction between NHERF1 and cpERM proteins.6 The cpERM–NHERF1 interaction occurs only after LPA stimulation, which is completely abolished by the deletion of the C-terminal region containing the ERM-binding domain (NHERF1-ΔCT).

NHERF1 translocation depends on the molecular interaction with ERM proteins phosphorylated at the C terminus (cpERMs) in response to LPA stimulation. (a) Lysophosphatidic acid (LPA)-induced ERM phosphorylation. OVCAR-3 cells were serum-deprived for 24 h, and then stimulated with LPA in various concentrations indicated. The levels of cpERM and the total amounts of ERM and NHERF1 were determined by western blotting using antibodies specific for cpERM, ERM proteins (ERM) or NHERF as indicated. (b) LPA-induced interaction between NHERF1 and cpERM proteins. FLAG-tagged NHERF1 wild-type (FLAG-NHERF1 WT) and a C-terminally deleted form (FLAG-NHERF1 ΔCT) were transfected into OVCAR-3 cells. After serum deprivation for 24 h, cells were stimulated with 1 μM LPA and lysed. FLAG-tagged NHERF1 was immunoprecipitated with anti-FLAG affinity resin. Co-immunoprecipitation of cpERM with FLAG-NHERF1 was monitored by western blotting using anti-cpERM antibody. (c, d) CpERM-dependent translocation of NHERF1. OVCAR-3 cells on collagen-coated cover glass were transfected with GFP-tagged NHERF1 constructs, either wild-type (NHERF1-WT) (c) or a mutant lacking the C-terminal ERM-binding region (NHERF1-ΔCT) (d). Serum-deprived cells were treated with vehicle or 1 μM LPA for the indicated times. After fixation and permeabilization, the cells were immunolabeled with anti-cpERM antibody. Time-dependent localization of both cpERM proteins and NHERF1 was examined under a confocal microscope with constant settings. Upon LPA stimulation, the cpERM level was markedly elevated, and cpERM co-localized at the cell margin and in surface protrusions with wild-type NHERF1 (c, arrows), but not with NHERF1-ΔCT (d, arrowheads). Scale bars, 20 μm. Relative fluorescence intensities are profiled along the line of the long arrow (blue) overlaid on each image, using the line scan tool of Image J software. The relative distributions of NHERF1 and cpERM proteins along the line (blue) are plotted in the graph (right panel of each image). (e, f) cpERM-specific co-localization of NHERF1. OVCAR-3 cells expressing EGFP-tagged NHERF1 were prepared as described above and immunostained with antibodies specific for C-terminal phosphorylated ERM proteins (e) or an anti-ERM antibody that preferentially binds unphosphorylated ERM proteins (f). Lower panels show higher magnifications of the yellow boxed regions of the upper panels. NHERF1 co-localized at protrusion structures with cpERM (e, arrows), but not with total ERM proteins (f, arrowheads). These results are representative of three independent experiments. Scale bars, 20 μm.
Next, we examined the significance of the NHERF1–ERM interaction in the translocation event. Wild-type NHERF1 (GFP-NHERF1 WT) translocated rapidly from the cytosol to the cell margin where cpERM proteins are generated, and subsequently to surface protrusions along with cpERM proteins (Figure 2c). After LPA stimulation, the localizations of the two proteins at the plasma membrane completely overlapped, especially at the cortical structure (Figure 2c). More importantly, NHERF1 translocation was completely abrogated by a C-terminal deletion mutation in NHERF1 (NHERF1-ΔCT) that resulted in loss of the ERM interaction (Figure 2d), demonstrating that the interaction with ERM proteins is essential for this translocation event. Consistent with this, high-magnification images revealed that NHERF1 co-localized with cpERM proteins at heavy protrusions in LPA-stimulated cells (Figure 2e). To confirm the significance of ERM phosphorylation in NHERF1 localization, we examined the subcellular localization of all ERM proteins using another ERM antibody that preferentially recognizes non-phospho-ERM proteins. Even in LPA-stimulated cells, non-phosphorylated ERM proteins exist in a subcortical layer beneath the plasma membrane, presumably through PIP2 interaction as the dormant forms (Figure 2f, arrowheads).46 It is obvious that NHERF1 (green, arrow) did not significantly overlap with the dormant ERM proteins (red, arrowheads), suggesting that NHERF1 and cpERM proteins engage in a specific interaction at the cortical structure. These results indicated that LPA-triggered ERM phosphorylation is responsible for rapid translocation of NHERF1 to the cortical structure.
NHERF1 potentiates ERM phosphorylation in LPA-stimulated cells by stabilizing cpERM proteins
We next attempted to determine the potential role of NHERF1–cpERM interaction in ERM regulation at the plasma membrane. To deplete NHERF1 proteins, we transfected NHERF1-specific siRNA duplex into OVCAR-3 cells (Figure 3a–d). We then investigated whether the absence of NHERF1 influences the phosphorylation status of ERM proteins in LPA-stimulated cells. NHERF1 depletion significantly decreased the LPA-induced level of ERM phosphorylation at all treatment times examined (Figure 3a). This inhibitory effect of NHERF1 depletion was reproduced in cells stimulated with various concentrations of LPA, whereas NHERF1 depletion had little effect on other LPA-triggered signal events such as phosphorylation of AKT, GSKα/β and ERK, suggesting that NHERF1 plays a specific role in the regulation of ERM proteins (Figure 3b).

NHERF1 potentiates ERM phosphorylation in LPA-stimulated cells. (a, b) Effect of NHERF1 depletion on LPA-induced ERM phosphorylation. OVCAR-3 cells were transfected with siRNA duplexes specific for NHERF1, NHERF2 or control siRNA (luciferase GL3). One day after transfection, the cells were re-plated onto collagen-coated culture dishes. One day later, the cells were serum-deprived for 24 h and treated with 1 μM LPA for various times (a) or with various doses of LPA for 10 min (b) as indicated. LPA-induced levels of cpERM, pAKT, pSGKα/β and pERK in the cell lysates were determined by western blot analysis with the indicated phospho-specific antibodies (a, b). The amounts of total ERM proteins, NHERF1 and NHERF2 in the same cell lysates were also determined using the appropriate cognate antibodies (a). Black arrows indicate cognate target proteins, and the blue arrowhead indicates nonspecific bands in the western blot image (a). These results are representative of three independent experiments. The relative intensity of each immunoblot image is shown in the graph at the right side. Data represent mean ±s.e.m. *P<0.05 and **P<0.01, t-test. (c) Expression levels of NHERF1 and ERM proteins in ovarian cell lines derived from either normal or cancerous epithelium. NHERF1 and ERM protein levels in the cell lysates of HIO-80, SK-OV-3 and OVCAR-3 are shown together with the GAPDH level as an input control. (d, e) Potentiation of ERM phosphorylation by overexpression of NHERF1. HIO-80 cells were transfected with GFP-NHERF1 (WT) or its C-terminally deleted form (ΔCT). Immunoblot analyses with anti-GFP and anti-NHERF1 antibody were performed to determine the expression level of each NHERF1 construct and the degree of overexpression of GFP-NHERF1 relative to the endogenous NHERF1 level, respectively (d). LPA-induced levels of cpERM and pERK, as well as total ERM level, were determined as described above. All of these results represent three independent experiments.
Conversely, we examined the effect of NHERF1 overexpression on the cpERM level following LPA stimulation. Despite the similarity in their tissue origin, HIO-80 cells (human immortalized ovary epithelial cell line) express a much lower level of NHERF1 than ovarian carcinoma cell lines (OVCAR-3 and SK-OV-3), without any significant difference in the level of ERM proteins (Figure 3c). Overexpression of GFP-tagged NHERF1 over the endogenous level significantly increased the LPA-induced level of cpERM proteins, whereas C-terminal deletion of NHERF1 abolished this effect completely, further confirming the importance of the direct interaction of NHERF1 with cpERM proteins for this regulation. Consistent with this, the effect of NHERF1 overexpression is selective for cpERM proteins and does not influence other phosphorylation events, as demonstrated for the pERK pathway (Figure 3d and e). These results demonstrate that, in response to LPA stimulation, NHERF1 is recruited to cpERM proteins, which stabilize and/or potentiate its activation status.
The NHERF1–cpERM complex is required for LPA-induced reorganization of the cell cortex
ERM proteins were initially identified as enriched in various membrane structures such as microvilli, membrane ruffles and filopodia.5, 19 In these structures, they play an essential role in actin cytoskeleton reorganization, assembly of focal adhesion complexes, and membrane dynamics by linking membrane proteins to the cortical actin cytoskeleton. Based on our observation of reciprocal regulation between NHERF1 and cpERM proteins, we hypothesized that the NHERF1–cpERM complex mediates LPA-induced dynamic membrane morphogenesis of OVCAR-3 cells. To test this idea, we overexpressed a phosphorylation-defective T567A mutant of Ezrin, which has a dominant-negative effect on endogenous ERM proteins. Overexpressed WT Ezrin was targeted to membrane protrusions with nearly the same localization pattern as cpERM proteins, whereas Ezrin-T567A was not (Figure 4a). Moreover, Ezrin-T567A overexpression markedly attenuated proper targeting of endogenous cpERM proteins to the cortical membrane and interfered with membrane protrusion formation elicited by LPA. This result indicated that cpERM proteins play pivotal roles in LPA-induced membrane reorganization/remodeling.

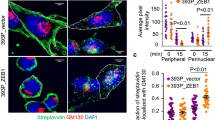
NHERF1 is required for cpERM-dependent dynamic reorganization of the cell cortex. (a) The role of ERM phosphorylation in LPA-induced remodeling of the cell cortex. OVCAR-3 cells were transfected with VSVG-tagged Ezrin constructs, including wild-type Ezrin and the phosphorylation-defective mutant (Ezrin-T567A). The transfected cells were treated with 1 μM LPA for 10 min. The fixed cells were immunostained with α-VSVG epitope and α-cpERM antibody, and then observed by confocal microscopy with the focus adjusted to either the top or the bottom side of the cells. CpERM proteins co-localized with VSVG-tagged Ezrin wild-type (arrows), but not with its dominant-negative mutant Ezrin-T567A (arrowheads). All images are from a single experiment, which was representative of three independent experiments. Scale bars, 20 μm. (b–d) Effect of NHERF1 depletion on cpERM-dependent reorganization of the cell cortex. OVCAR-3 cells were transfected with siRNA duplexes specific for NHERF1 or −2. The transfected cells were re-plated onto collagen-coated glass coverslips and serum-deprived for 24 h prior to 1 μM LPA treatment for 10 min. Generation and subcellular localization of cpERM protein was visualized by immunostaining with anti-cpERM antibody (b). Images were acquired by confocal microscopy with the focus adjusted to either the top or the bottom side of the cells. CpERM localization is indicated at surface protrusions (arrows) and at the cell margin (arrowheads). All images are from a single experiment that was representative of three independent experiments. Scale bars, 20 μm. Specific gene silencing of NHERF1 and NHERF2 with transfection with isoform-specific siRNAs (c). Protein levels of NHERF1 and NHERF2 were determined using specific antibodies. Representative western blot images for each group and NHERF isoform are shown. Proportion of LPA-stimulated cells with heavy protrusions (d). Images in each slide were captured in 5–7 randomly selected high-power fields (HPF, × 20 objective) with constant settings. Number of total cells and cells with heavy protrusions defined as those with typical protrusive structures covering the cell edge of the bottom region by more than 1/4. Representative images of the cells with or without typical heavy protrusions are as shown in c. The results from each slide show the percentage of the cells with heavy protrusions. Each bar represents the mean±s.e.m. from three independent experiments. All data represent mean±s.e.m. Pair-wise post hoc test, #P<0.001, *P<0.05, NS, non-significant.
We next examined the effect of NHERF1 depletion on LPA-induced reorganization of the cell cortex. CpERM proteins in control cells were targeted to surface protrusion structures generated in either the membrane surface of the top region or the cell margin of the bottom region. By contrast, in NHERF1-deficient cells, cpERM proteins were present at markedly lower levels, and the cells were impaired in generation of surface protrusions (Figure 4b). This effect was marginal in NHERF2-deficient cells, suggesting that NHERF1 makes a major contribution to cpERM regulation in OVCAR-3 cells. Quantitative analysis of the cells with heavy protrusions revealed that NHERF1 knockdown caused marked reduction in LPA-induced formation of surface protrusion structures (Figure 4c and d). These results strongly support the functional significance of NHERF1–cpERM protein complexes in the regulation of dynamic membrane morphogenesis.
NHERF1–cpERM complexes are enriched in pseudopodia of OVCAR-3 cells migrating toward a LPA gradient
During migration, tumor cells form a distinct plasma membrane domain, the pseudopod, whose generation and stabilization determines the directionality of cell movement.47 The rapid movement of cells requires local dynamics in the cytoskeleton and membrane; consequently, cell motility requires a supply of the appropriate molecular machinery at the site of pseudopodial extension. In light of the LPA-dependent regulation of the NHERF1–cpERM complex and its crucial role in membrane morphogenesis, we explored the possibility that the NHERF1–cpERM complex is targeted to the pseudopod, where dynamic remodeling in membrane morphology happens toward chemotactic stimulus. To visualize NHERF1 and cpERM proteins in the pseudopodia, we immunostained cells that were fixed during migration through a transparent porous membrane. In these experiments, OVCAR-3 cells were mounted on the upper side of the porous membrane, and the reservoir on the lower side was filled with migration buffer containing 1 μM LPA; the cells were then allowed to extend pseudopodia through the membrane toward the LPA stimulus (Figure 5a, left panel). We modified our chemotaxis assay by decreasing both the pore size of the membrane (5 μm) and the incubation time (1 h); these conditions allow pseudopodial projection, but not migration of the cell body (Figure 5a, left panel). In this model, cpERM proteins were highly enriched in pseudopodia only when LPA was present in the lower chamber (Figure 5aa–c). We further validated the pseudopodial localization of cpERM proteins at the underside of the porous membrane by nuclear staining of the cell body on the upper side (Figure 5ab), as well as by removing any residual cells on the upper side following chemotaxis (Figure 5ac). At higher magnification, we observed that cpERM proteins were targeted to protrusive structures of the pseudopodia (Figure 5ba and -b), but to a much lesser extent to the cell membrane near the cell body, suggesting uneven activation of ERM proteins toward the LPA gradient. Notably, this cpERM localization was overlapped with that of NHERF1 not only in the pseudopodia, but also in their protrusion structures (Figure 5c and b and -c), but not with control GFP protein (Figure 5ca). Collectively, these results indicated that NHERF1 translocates to sites on the membrane where cpERM proteins are generated in the direction of the LPA gradient, leading to pseudopodial enrichment of the NHERF1–cpERM protein complex, followed by local membrane morphogenesis involved in chemotactic cell migration.

NHERF1/cpERM complex is highly enriched at pseudopodia of cells migrating toward a LPA gradient. (a) Localization of cpERM proteins at the pseudopodia of migrating cells. Schematic representation of pseudopodia formation of cells migrating toward a LPA gradient (left panel). OVCAR-3 cells were mounted onto collagen-coated 5 μm porous membranes in modified Boyden chambers, and then allowed to extend pseudopodia toward the bottom chamber in the absence (aa) or the presence (ab) of LPA (1 μM) for 1 h. In addition, to confirm pseudopodia formation on the lower surface of the porous membrane, cell bodies on the upper side were cleared with a cotton swab (ac). Cell preparation is illustrated schematically above each set of images (aa–c). In fixed cells, the nucleus in the cell body and cpERM in pseudopodia were visualized with PI (propidium iodide) staining and immunostaining with anti-cpERM antibody/Alexa Fluor 488-conjugated secondary antibody, respectively. The plane of focus of confocal microscopy was sequentially adjusted to the top membrane surface (upper) and then the lower membrane surface (lower) in the same field. All results are from a single experiment that was representative of three experiments performed on independent preparations. Scale bars, 20 μm. (b) CpERM localization at the protrusion structures at the pseudopodia. Migratory OVCAR-3 cells were prepared as illustrated above each image set. To determine the subcellular location of cpERM in the migratory cells at the higher resolution (× 40 objective), the plane of focus of confocal microscopy was sequentially adjusted to the upper cell body (Upper), porous membrane (Memb.) and lower pseudopodia (Lower) in a single cell, as illustrated in the left panel. CpERM proteins are enriched in protrusive structures of the pseudopodia (arrow in ba/b), and to a much lesser extent in the cell body (arrowhead in ba). Scale bars, 10 μm. (c) Co-localization of NHERF1 with cpERM at pseudopodia of migrating cells. OVCAR-3 cells transfected with either GFP-NHERF1 or GFP control vector were mounted onto the porous membrane of a modified Boyden chamber and prepared as illustrated above each set of images. GFP-NHERF1 and cpERM proteins were visualized in multiple planes of focus, which were sequentially adjusted to the upper cell body, porous membrane and lower pseudopodia in a single cell as indicated. CpERM co-localized at the protrusion of the pseudopodia with GFP-NHERF1 (arrows in cb/c), but not with control GFP (arrowheads in ca). All these results are from a single experiment that was representative of three experiments performed with independent preparations. Scale bars, 10 μm.
NHERF1 plays a pivotal role in LPA-induced cell migration of gynecological carcinoma cells
Based on observation of dynamic targeting of the NHERF1–cpERM complex to the pseudopodia, we postulated that this complex is required for the directional migration of tumor cells toward a LPA gradient. First, we examined the effect of NHERF1 truncation mutant expression on pseudopodia formation (Figure 6a and b). In contrast to full-length NHERF1 (NHERF1-WT, left panel), the C-terminal fragment (NHERF1-CT, right panel) completely abrogated pseudopodia formation in the majority of the transfected cells, suggesting that its potent dominant-negative activity was mediated via its competitive interference with the NHERF1–cpERM interaction. In addition, a C-terminal deletion of NHERF1 (NHERF1-ΔCT, middle panel) inhibited pseudopodia formation, but much less strongly than NHERF1-CT. Nonetheless, pseudopodia formation was significantly attenuated in cells expressing NHERF1-ΔCT at high levels (arrows), but not in the cells with low or undetectable expression (yellow arrowheads). Importantly, both NHERF1 mutants exerted the same inhibitory effects on pseudopodia formation, suggesting that full-length NHERF1-dependent molecular scaffolding established by both the two N-terminal PDZ domains and the C-terminal cpERM-binding region may be required for pseudopodia formation for the migratory process.

NHERF1 is required for chemotactic migration of cancer cells. (a) Schematic domain structures of GFP-labeled NHERF1 constructs including the wild type (WT), C-terminally truncated form (ΔCT), and C-terminal fragment (CT) (left panel). Expression levels of GFP-NHERF1 constructs were determined using anti-GFP antibody (right panel) as indicated by the arrows. (b) Inhibition of pseudopodia formation by overexpression of NHERF1 deletion mutants. OVCAR-3 cells were transfected with GFP-NHERF1 constructs, and migration assays were performed 2 days after transfection, as described above. The migratory cells fixed on the membrane were probed with anti-cpERM antibody. Pseudopodial localizations of GFP-NHERF1 and cpERM proteins were imaged under confocal microscopy with constant settings. These results are from a single experiment that was representative of three experiments performed on independent preparations. Scale bar, 20 μm. (c) siRNA-mediated silencing of NHERF1 expression in various cell lines derived from gynecological cancers. OVCAR-3 (ascites-derived ovarian cancer cell line), SK-OV-3 (ascites-derived ovarian cancer cell line) and MDA-MB-231 cells (breast carcinoma cell line) were tested in this study. Each cell line was transfected with either siRNA duplex specific for NHERF1 or control siRNA. NHERF1 levels in cell lysates were determined by western blotting with anti-NHERF1 antibody. (d) Effect of NHERF1 depletion on LPA-induced migration of OVCAR-3 cells. Three days after siRNA transfection, OVCAR-3 cells were mounted onto a collagen-coated porous membrane (pore size, 8 μm) of a modified Boyden chamber and allowed to migrate to the lower side for 3.5 h in the presence or absence of 1 μM LPA in the lower chamber. Migratory cells in the lower membrane were visualized by Hoechst 33342 staining of nuclei. These images from OVCAR-3 cells represent three independent experiments. (e) Effect of NHERF1 depletion on the migration of gynecological cancer cell lines. OVCAR-3, SK-OV-3 and MDA-MB-231 cells were tested in this study. Each cell line was transfected with siRNA duplexes as described in (c). After chemotactic migration assays, the membranes were observed by fluorescence microscopy, and images were captured in five randomly chosen high-power fields (HPF, × 20 objective). The numbers of migratory cells per membrane were directly counted and analyzed statistically in each cell line. Each bar represents the mean±s.e.m. from three independent experiments. All data represent mean ±s.e.m. *P<0.05 and NS, non-significant.
Based on our observation of LPA-induced assembly of the NHERF1–cpERM complex at polar pseudopodia, we investigated whether this molecular complex mediates the chemotactic migration of those cancer cells toward LPA. First, we observed that the phosphorylation-defective ezrin mutant (Ezrin-T567A) blocked LPA-induced migration of OVCAR-3 cells (Supplementary Figure 1A–C). Consistent with this, in gynecological cancer cell lines such as OVCAR-3, SK-OV-3, and MDA-MB-231, NHERF1 depletion resulted in the almost complete loss of migration toward the LPA gradient (Figure 6c–e). Collectively, those results suggested that cytosolic NHERF1 plays a significant role in directional migration of gynecological cancer cells in response to LPA.
Discussion
The classical apical scaffolding protein NHERF1 was previously shown to relocalize to the cytoplasm in post-EMT cancer cells, but its role in tumor progression remained unclear. Here we showed that cytosolic NHERF1 dynamically translocates to the plasma membrane upon LPA stimulation of ascites-derived OVCAR-3 ovarian cancer cells. Translocation of NHERF1 is mediated through molecular interaction with cpERMs at the plasma membrane of LPA-stimulated cells (Figure 7a and b). cpERM and NHERF1 are engaged in reciprocal regulation, and form a molecular complex that plays a significant role in dynamic remodeling of the cell cortex (Figure 7c and d). In vitro chemotactic migration assays revealed that the NHERF1–cpERM complex is highly enriched in a frontal pseudopodial structure extending toward the LPA gradient that is essential for cancer cell migration (Figure 7e). Our results suggest that local translocation of cytoplasmic NHERF1 provides post-EMT cancer cells with a means to freely move toward a chemotactic stimulus (in this case, LPA). Taken together, these results suggested that NHERF1 dynamics play a significant role in controlling cancer cell behavior in the tumor microenvironment, and likely promote ovarian cancer progression.

Schematic representation of LPA-induced NHERF1 regulation and its implication for cancer progression. (a) In the resting state, dormant ERM proteins, which exist as inactive monomers or oligomers, localize primarily to the plasma membrane via interaction with PIP2. (b) Upon LPA stimulation, ERM proteins are rapidly phosphorylated at the C terminus and undergo a phosphorylation-induced transition to active monomers at the plasma membrane. (c) NHERF1 is recruited to membrane-anchored cpERM proteins through the ERM-binding region at the NHERF1 C terminus. (d) The membrane-targeted NHERF1–cpERM complex stabilizes ERM phosphorylation to play a key role as a molecular scaffold, tethering multiple membranous and cytosolic partners just beneath the plasma membrane, which leads to the dynamic reorganization of the cell cortex. (e) Human epithelium is composed of cellular monolayers in which individual cells are tightly linked to their neighbors by tight junctions. In normal epithelium, NHERF1 localizes exclusively in the apical microvilli of the polarized epithelial cells and plays broad roles ranging from regulation of apical proteins to morphogenesis of the apical membrane itself. However, during the epithelial–mesenchymal transition (EMT), innately polar cells lose the cell-to-cell junctions that are required for epithelial integrity and thus lose intrinsic membrane polarity. Eventually, these post-EMT cells behave in an ‘amoeboid’ manner, and are capable of freely moving in any direction. Here we propose a working model in which cytosolic NHERF1 in cancerous amoeboid cells can be dynamically redistributed toward extracellular stimuli via interaction with cpERM, and play a key role in the chemotactic cell migration by resetting the front–rear polarity.
Reciprocal regulation of NHERF1 and cpERM proteins
We showed that NHERF1 translocation depends on LPA-induced ERM phosphorylation and subsequent interaction via the NHERF1 C terminus underneath the plasma membrane, validating the important role of ERM proteins in NHERF1 regulation in cancer cells, as in normal epithelial cells. In dormant monomer of the ERM family proteins, N-ERMAD (ERM-association domains) for binding site of membrane-associated proteins binds very tightly to its C-terminal F-actin-binding site, C-ERMAD in the closed conformation.7 Activation of ERM proteins to unmask their binding sites involves dissociation of the intramolecular N-/C-ERMAD interaction, which is achieved by two sequential steps: PIP2 binding to the N-terminal domains, and subsequent phosphorylation of a C-terminal threonine residue by multiple cellular kinases.3 It is noteworthy that, even before extracellular stimulation, most non-phosphorylated ERM proteins localize throughout the inner face of the plasma membrane (Figure 2f), consistent with the altered PI(4,5)P2 localization in post-EMT cells.48 Membrane-bound ERM proteins still remain poorly activated even when PIP2 is bound to their N-terminal domains, but are prone to become robustly activated in response to LPA stimulation, which triggers their C-terminal phosphorylation. Eventually, cpERM proteins become sufficiently active to recruit NHERF1 to the plasma membrane. Our current observations demonstrate the significant role of cpERM proteins not only in static localization of NHERF1 at the normal epithelium,5, 8 but also in active translocation of NHERF1 in post-EMT cells, suggesting that the cpERM–NHERF1 complex is preserved both before and after the EMT, despite the significant difference in its physiological and pathological roles in each context.
In addition to demonstrating cpERM-dependent regulation of NHERF1 localization, we showed that NHERF1 is a key regulator of cpERM proteins. NHERF1 depletion significantly decreased the cpERM level and abolished cpERM-dependent cortical reorganization (Figure 3a and b). Presumably, NHERF1 could influence the cpERM level through either potentiation of ERM phosphorylation or stabilization of cpERMs. It is well established that NHERF1 selectively binds to cpERMs, but not to non-phospho-ERMs.45, 46 Accordingly, our immunocytochemistry revealed that NHERF1 co-localized with cpERMs, but not with non-phospho-ERM proteins. Notably, NHERF1 depletion decreased cpERM levels, but did not abolish phosphorylation itself (Figures 3a and b, and 4b). Moreover, NHERF1 depletion did not influence other major phosphorylation events in the AKT, ERK1/2 and SGKα/β pathways (Figure 3b), suggesting selective regulation of cpERMs by NHERF1, possibly mediated by direct interactions. This line of evidence supports the idea that NHERF1 regulates the stability of cpERMs in a protein complex, rather than acting upstream of ERM phosphorylation. Consistent with this, several studies described the rapid dynamics of ERM phosphorylation.10, 49 Estimation of the rate of Ezrin phosphorylation and dephosphorylation using phosphatase and kinase inhibitors revealed that Ezrin undergoes a phosphocycle of ~1–2 min in normal epithelial cells. If phosphocycling is suppressed by inhibiting dephosphorylation, the polarized distribution of Ezrin in epithelial cells is lost,3, 50 suggesting that phosphocycling of ERM proteins is constitutive, probably due to uneven subcellular distribution of kinases, phosphatases and other regulatory factors such as NHERF1 within various cellular compartments. In this regard, NHERF1 interaction may protect cpERM from phosphatase-dependent dephosphorylation by masking the phosphosite directly. It is also plausible that the NHERF1–cpERM complex may isolate itself inside the cortical protrusion structure, away from the subcellular region enriched in phosphatases, during cortical reorganization (Figure 4b). However, we cannot exclude the possibility that NHERF1 works upstream of ERM phosphorylation. ERM proteins are phosphorylated by several cellular kinases, including Rho-associated kinase (ROCK),45 myotonic dystrophy-related Cdc42-binding kinases (MRCK),51 PKC-θ,52 and mammalian STE20-like protein kinase 4 (MST4)53, 54 in epithelial cells, and lymphocyte-oriented kinase (LOK)/STE20-like kinase (SLK) in lymphocyte and Caco2 cells.50, 55 We have yet to examine the expression patterns of those kinases in OVCAR-3 cells or the potential influence of NHERF1 depletion on those kinase activities upon LPA stimulation. Moreover, cpERM/NHERF1 may recruit several Rho family GTPase regulators such as RhoGDI and RhoGEF (Dbl) into the functional complex,7, 45 which may provide a positive feedback loop in the LPA-induced RhoGTPase pathway and ultimately potentiate ERM phosphorylation. On the other hand, previous studies have shown that NHERF1 is a substrate for diverse cellular kinases whose phosphorylation of NHERF1 regulates its self-oligomerization and protein interactions with other PDZ ligands.10, 56, 57, 58, 59, 60 We are not yet able to determine if NHERF1 is phosphorylated together with other ERM proteins upon LPA stimulation. If this turns out to be the case, then NHERF phosphorylation could be engaged in further crosstalk with ERM phosphorylation, resulting in reciprocal regulation of their activities. Although the mechanistic regulation model needs to be further clarified, it is clear that NHERF1 and cpERM engage in reciprocal regulation within a functional complex during LPA-triggered signaling events in cancer cells.
The NHERF family of protein has four members.17 Among them, NHERF1 shares with NHERF2 52% amino-acid identity, a conserved domain architecture, and common ligands.14 NHERF1 and NHERF2 both have extremely high affinities for the FERM domain of ERM proteins, and exhibit some overlap in PDZ ligand specificity and tissue distributions.61, 62 Recent reports highlighted the difference between NHERF1 and NHERF2 in terms of molecular dynamics and subcellular localizations. NHERF1 displays more active dynamics than NHERF2 due to subtle differences in its PDZ ligands and C-terminal ERM-binding domain.63, 64 In addition, NHERF1 predominantly localizes in microvilli, whereas NHERF2 localizes in intracellular submicrovilli and vesicular structures. In this regard, NHERF1 differs significantly from NHERF2 regarding its role in regulation of cpERM proteins and subsequent cortical reorganization (Figures 3a and b, and 4b–d), providing additional evidence that NHERF1 and NHERF2 play non-redundant roles. NHERF1 co-localizes with cpERM proteins in protrusion structures of OVCAR-3 ovarian cancer cells, as also observed in microvilli of normal epithelial cells. In this regard, distinct molecular interactions of NHERF1 with PDZ ligands and cpERMs may account for the unique role of NHERF1 in dynamic reorganization of the cell cortex in ovarian cancer cells.
Molecular dynamics of cytoplasmic NHERF1 and their role in ovarian cancer progression
Cell polarity is defined as asymmetric distribution of cellular components and their molecular complexes to particular regions within cells.65 This asymmetry, especially at the plasma membrane, is required to establish not only the apical–basal axis in normal epithelium, but also front–rear polarity in the mesenchymal cells.1 NHERF1 localizes exclusively underneath the apical membrane of normal epithelial layers and serves as a molecular scaffold to cluster various signaling molecules into stable complexes.5, 8 The restriction of NHERF1 localization is important for microvilli formation on the apical surface of epithelial cells.8, 10 In contrast to normal apical localization, NHERF1 mainly localizes in the cytoplasm of invasive carcinoma cells, often concomitant with overexpression.32, 34, 36, 38, 39, 40, 41, 66 This relocalization is strongly associated with cancer progression, metastatic potential, poor prognosis and estrogen responsiveness, especially in carcinomas. Furthermore, NHERF1 binds to tumor suppressors, including PTEN and beta-catenin, at the plasma membrane in normal epithelial cells.39, 40, 67 These observations have led to a mechanistic model in which NHERF1 acts as a tumor suppressor when it is localized at the plasma membrane, but as a pathogenic protein when it is localized in the cytoplasm of cancer cells.67 The mechanism underlying the functional transition of NHERF1 between normal epithelial cells and the cancer cell remains unknown.
Here we observed that cytoplasmic NHERF1 rapidly translocates to the plasma membrane upon LPA stimulation in ovarian cancer cells that have lost apical–basal polarity. More importantly, if the LPA stimulus is given unevenly, NHERF1 translocates asymmetrically to the frontal side, that is, toward the LPA gradient. This asymmetric membrane translocation of NHERF1 is crucial for chemotactic cell migration toward a LPA stimulus. Presumably, in return for the loss of stable apical localization, NHERF1 gains the freedom to move to any region on the plasma membrane, and thus the ability to actively reset the front–rear polarity in post-EMT cells, especially in response to extracellular stimuli as exemplified by LPA. Thus, cancer cells appear to hijack polarity factors, including NHERF1, for tumor progression by simply redirecting from the apical–basal polarity of normal epithelium to front–rear polarity in post-EMT cells. In the current study, we demonstrated that dynamic relocalization of cytoplasmic NHERF1 mediates the directional cell migration of ovarian cancer cells toward LPA stimuli. Our findings suggest that the high dynamics of cytosolic NHERF1 provide cancer cells with a means of controlling chemotactic migration, with ensuring maximum plasticity for determining the moving direction. We believe that our study contributes to understanding of the pathophysiological role of cytosolic NHERF1 in cancer cells with no fixed polarity.
Recently emerging evidence indicated that ERM proteins are closely involved in tumor development.68, 69 They are frequently overexpressed in human tumors derived from breast, ovary, and rhabdosarcoma, and abnormal distribution of these factors is correlated with poor prognosis in breast cancer patients.70, 71 Ezrin relocalizes from apical membranes in normal epithelium to the cytoplasm of breast tumors,71 and ERM proteins promote invasion by tumor cells.69, 72 The molecular dynamics of the cpERM–NHERF1 complex may be associated with directional cell migration, and with metastasis in cancer. In this context, our results reveal a novel mechanism by which cytoplasmic NHERF1 contributes to cancer progression, in accordance with the pathogenic role of ERM family proteins.
Ovarian cancers are the gynecological malignancies with the highest case fatality rate,73 ascribed to the fact that the majority of patients are diagnosed with wide metastasis within the peritoneal cavity.74 During tumor progression, ovarian cancer cells metastasize either by directly extending from the epithelial tumor mass into neighboring organs (bladder/colon) or by detaching from the primary tumor and disseminating throughout the peritoneum. Extensive seeding of the ovarian cancer cells within the peritoneal cavity is associated with ascites, particularly in the advanced malignancy.74 LPA has been implicated in ovarian cancer progression30, 75 and is highly enriched in ascites of most patients with malignant ovarian cancer,22, 25, 26, 27, 28 and expression of LPAR is aberrantly altered in ovarian cancer cells,29, 76 implicating the LPA/LPAR axis in poor prognosis of ovarian cancer, especially in cases with extensive metastasis into nearby tissue or the peritoneum.31, 75 In ovarian cancer cells, LPA can strongly stimulate multiple cellular responses such as cell proliferation, cell migration/invasion and metastasis.21, 22 Nonetheless, the intracellular mechanism underlying LPA-induced progression of ovarian cancer has remained unclear. Notably in this regard, our findings in this study reveal that the cpERM–NHERF1 complex is essential for LPA-triggered migration of ascites-derived OVCAR-3 ovarian cancer cells. In amoeboid cancer cells that have lost their polarity, NHERF1 displays abnormal intracellular behavior with dynamic shuttling between cytoplasm and membrane toward the extracellular LPA gradient. This provides the cancer cells with the ability to engage in chemotactic migration and metastasis. Considering the pathological significance of LPA in ovarian cancer progression, molecular intervention in the LPA–cpERM–NHERF1 pathway should be subjected to further in vivo study to determine whether inhibition of this pathway could be used to prevent or treat ovarian cancer metastasis.
Now we have to admit that our current study is yet to prove in vivo relevance of LPA–cpERM–NHERF1 pathway by using either cancer patient-derived tissue or the xenograft model for ovarian carcinoma. Practically, it does not seem easy to follow these phosphorylation-dependent events in human carcinoma tissues that are generally processed without any specific treatment to preserve phospho-proteins and their protein complexes to truly reflect those found at the tumor mass inside the body. In addition, the various xenograft models have been developed mainly through the intraperitoneal injection of ovarian cancer-derived cell lines. Those models have been used to elucidate molecular events that regulate the ‘terminal transition’ from free-floating ascetic cells to metastatic lesions anchored on the peritonieum.77 Unfortunately, those models rarely contribute to our understanding of the very earliest events in the metastasis of ovarian cancers, such as the migration of tumor cells with metastatic potentials toward the intraperitoneal cavity and the shedding of cells from the primary tumor mass.77 In this regard, future studies using advanced experimental model are warranted to further verify in vivo relevance of LPA–cpERM–NHERF1 pathway as well as its pathological importance for the progression of ovarian cancer.
References
Lamouille S, Xu J, Derynck R . Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014; 15: 178–196.
Donowitz M, Cha B, Zachos NC, Brett CL, Sharma A, Tse CM et al. NHERF family and NHE3 regulation. J Physiol 2005; 567: 3–11.
Sauvanet C, Wayt J, Pelaseyed T, Bretscher A . Structure, regulation, and functional diversity of microvilli on the apical domain of epithelial cells. Annu Rev Cell Dev Biol 2015; 31: 593–621.
Shenolikar S, Weinman EJ . NHERF: targeting and trafficking membrane proteins. Am J Physiol Renal Physiol 2001; 280: F389–F395.
Viswanatha R, Bretscher A, Garbett D . Dynamics of ezrin and EBP50 in regulating microvilli on the apical aspect of epithelial cells. Biochem Soc Trans 2014; 42: 189–194.
Reczek D, Berryman M, Bretscher A . Identification of EBP50: a PDZ-containing phosphoprotein that associates with members of the ezrin-radixin-moesin family. J Cell Biol 1997; 139: 169–179.
Fehon RG, McClatchey AI, Bretscher A . Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol 2010; 11: 276–287.
Morales FC, Takahashi Y, Kreimann EL, Georgescu MM . Ezrin-radixin-moesin (ERM)-binding phosphoprotein 50 organizes ERM proteins at the apical membrane of polarized epithelia. Proc Natl Acad Sci USA 2004; 101: 17705–17710.
Saotome I, Curto M, McClatchey AI . Ezrin is essential for epithelial organization and villus morphogenesis in the developing intestine. Dev Cell 2004; 6: 855–864.
Garbett D, LaLonde DP, Bretscher A . The scaffolding protein EBP50 regulates microvillar assembly in a phosphorylation-dependent manner. J Cell Biol 2010; 191: 397–413.
Hanono A, Garbett D, Reczek D, Chambers DN, Bretscher A . EPI64 regulates microvillar subdomains and structure. J Cell Biol 2006; 175: 803–813.
LaLonde DP, Garbett D, Bretscher A . A regulated complex of the scaffolding proteins PDZK1 and EBP50 with ezrin contribute to microvillar organization. Mol Biol Cell 2010; 21: 1519–1529.
Weinman EJ, Steplock D, Wang Y, Shenolikar S . Characterization of a protein cofactor that mediates protein kinase A regulation of the renal brush border membrane Na(+)-H+ exchanger. J Clin Invest 1995; 95: 2143–2149.
Yun CH, Oh S, Zizak M, Steplock D, Tsao S, Tse CM et al. cAMP-mediated inhibition of the epithelial brush border Na+/H+ exchanger, NHE3, requires an associated regulatory protein. Proc Natl Acad Sci USA 1997; 94: 3010–3015.
Seidler U, Singh AK, Cinar A, Chen M, Hillesheim J, Hogema B et al. The role of the NHERF family of PDZ scaffolding proteins in the regulation of salt and water transport. Ann NY Acad Sci 2009; 1165: 249–260.
Voltz JW, Weinman EJ, Shenolikar S . Expanding the role of NHERF, a PDZ-domain containing protein adapter, to growth regulation. Oncogene 2001; 20: 6309–6314.
Weinman EJ, Hall RA, Friedman PA, Liu-Chen LY, Shenolikar S . The association of NHERF adaptor proteins with g protein-coupled receptors and receptor tyrosine kinases. Ann Rev Physiol 2006; 68: 491–505.
Oh YS, Jo NW, Choi JW, Kim HS, Seo SW, Kang KO et al. NHERF2 specifically interacts with LPA2 receptor and defines the specificity and efficiency of receptor-mediated phospholipase C-beta3 activation. Mol Cell Biol 2004; 24: 5069–5079.
Bretscher A, Chambers D, Nguyen R, Reczek D . ERM-Merlin and EBP50 protein families in plasma membrane organization and function. Annu Rev Cell Dev Biol 2000; 16: 113–143.
Jung Kang Y, Su Jeon E, Jin Lee H, Oh YS, Suh PG, Sup Jung J et al. NHERF2 increases platelet-derived growth factor-induced proliferation through PI-3-kinase/Akt-, ERK-, and Src family kinase-dependent pathway. Cell Signal 2004; 16: 791–800.
Choi JW, Herr DR, Noguchi K, Yung YC, Lee CW, Mutoh T et al. LPA receptors: subtypes and biological actions. Annu Rev Pharmacol Toxicol 2010; 50: 157–186.
Mills GB, Moolenaar WH . The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer 2003; 3: 582–591.
Mutoh T, Rivera R, Chun J . Insights into the pharmacological relevance of lysophospholipid receptors. Br J Pharmacol 2012; 165: 829–844.
Imamura F, Horai T, Mukai M, Shinkai K, Sawada M, Akedo H . Induction of in vitro tumor cell invasion of cellular monolayers by lysophosphatidic acid or phospholipase D. Biochem Biophys Res Commun 1993; 193: 497–503.
Xu Y, Gaudette DC, Boynton JD, Frankel A, Fang XJ, Sharma A et al. Characterization of an ovarian cancer activating factor in ascites from ovarian cancer patients. Clin Cancer Res 1995; 1: 1223–1232.
Baker DL, Morrison P, Miller B, Riely CA, Tolley B, Westermann AM et al. Plasma lysophosphatidic acid concentration and ovarian cancer. JAMA 2002; 287: 3081–3082.
Okita M . Abnormal plasma lysophosphatidic acid level in ovarian cancer patients. Bull Facul Health Welfare Sci 1994; 1: 29–35.
Xu Y, Shen Z, Wiper DW, Wu M, Morton RE, Elson P et al. Lysophosphatidic acid as a potential biomarker for ovarian and other gynecologic cancers. JAMA 1998; 280: 719–723.
Jahn SC, Law ME, Corsino PE, Parker NN, Pham K, Davis BJ et al. An in vivo model of epithelial to mesenchymal transition reveals a mitogenic switch. Cancer Lett 2012; 326: 183–190.
Willier S, Butt E, Grunewald TGP . Lysophosphatidic acid (LPA) signalling in cell migration and cancer invasion: a focussed review and analysis of LPA receptor gene expression on the basis of more than 1700 cancer microarrays. Biol Cell 2013; 105: 317–333.
Murph MM, Liu W, Yu S, Lu Y, Hall H, Hennessy BT et al. Lysophosphatidic acid-induced transcriptional profile represents serous epithelial ovarian carcinoma and worsened prognosis. PLoS ONE 2009; 4: e5583.
Wang L, Du YR, Ji MY, Wang W, Zhan N, Zhou QS et al. Reduced EBP50 expression or mis-localization of the EBP50 protein is associated with the malignant progression of esophageal squamous cell carcinoma. Eur Rev Med Pharmacol Sci 2014; 18: 3854–3863.
Dai JL, Wang L, Sahin AA, Broemeling LD, Schutte M, Pan Y . NHERF (Na+/H+ exchanger regulatory factor) gene mutations in human breast cancer. Oncogene 2004; 23: 8681–8687.
Cardone RA, Bellizzi A, Busco G, Weinman EJ, Dell'Aquila ME, Casavola V et al. The NHERF1 PDZ2 domain regulates PKA-RhoA-p38-mediated NHE1 activation and invasion in breast tumor cells. Mol Biol Cell 2007; 18: 1768–1780.
Schindelmann S, Windisch J, Grundmann R, Kreienberg R, Zeillinger R, Deissler H . Expression profiling of mammary carcinoma cell lines: correlation of in vitro invasiveness with expression of CD24. Tumour Biol 2002; 23: 139–145.
Song J, Bai J, Yang W, Gabrielson EW, Chan DW, Zhang Z . Expression and clinicopathological significance of oestrogen-responsive ezrin-radixin-moesin-binding phosphoprotein 50 in breast cancer. Histopathology 2007; 51: 40–53.
Bellizzi A, Greco MR, Rubino R, Paradiso A, Forciniti S, Zeeberg K et al. The scaffolding protein NHERF1 sensitizes EGFR-dependent tumor growth, motility and invadopodia function to gefitinib treatment in breast cancer cells. Int J Oncol 2015; 46: 1214–1224.
Hayashi Y, Molina JR, Hamilton SR, Georgescu MM . NHERF1/EBP50 is a new marker in colorectal cancer. Neoplasia 2010; 12: 1013–1022.
Kreimann EL, Morales FC, de Orbeta-Cruz J, Takahashi Y, Adams H, Liu TJ et al. Cortical stabilization of beta-catenin contributes to NHERF1/EBP50 tumor suppressor function. Oncogene 2007; 26: 5290–5299.
Takahashi Y, Morales FC, Kreimann EL, Georgescu MM . PTEN tumor suppressor associates with NHERF proteins to attenuate PDGF receptor signaling. EMBO J 2006; 25: 910–920.
Saponaro C, Malfettone A, Dell'Endice TS, Brunetti AE, Achimas-Cadariu P, Paradiso A et al. The prognostic value of the Na(+)/ H(+) exchanger regulatory factor 1 (NHERF1) protein in cancer. Cancer Biomark 2014; 14: 177–184.
Zhang WW, Fang X, Mazur W, French BA, Georges RN, Roth JA . High-efficiency gene transfer and high-level expression of wild-type p53 in human lung cancer cells mediated by recombinant adenovirus. Cancer Gene Ther 1994; 1: 5–13.
Chen HC . Boyden chamber assay. Methods Mol Biol 2005; 294: 15–22.
Viswanatha R, Wayt J, Ohouo PY, Smolka MB, Bretscher A . Interactome analysis reveals ezrin can adopt multiple conformational states. J Biol Chem 2013; 288: 35437–35451.
Matsui T, Maeda M, Doi Y, Yonemura S, Amano M, Kaibuchi K et al. Rho-kinase phosphorylates COOH-terminal threonines of ezrin/radixin/moesin (ERM) proteins and regulates their head-to-tail association. J Cell Biol 1998; 140: 647–657.
Fievet BT, Gautreau A, Roy C, Del Maestro L, Mangeat P, Louvard D et al. Phosphoinositide binding and phosphorylation act sequentially in the activation mechanism of ezrin. J Cell Biol 2004; 164: 653–659.
Nabi IR . The polarization of the motile cell. J Cell Sci 1999; 112 (Pt 12): 1803–1811.
Shewan A, Eastburn DJ, Mostov K . Phosphoinositides in cell architecture. Cold Spring Harbor Perspect Biol 2011; 3: a004796.
Coscoy S, Waharte F, Gautreau A, Martin M, Louvard D, Mangeat P et al. Molecular analysis of microscopic ezrin dynamics by two-photon FRAP. Proc Natl Acad Sci USA 2002; 99: 12813–12818.
Viswanatha R, Ohouo PY, Smolka MB, Bretscher A . Local phosphocycling mediated by LOK/SLK restricts ezrin function to the apical aspect of epithelial cells. J Cell Biol 2012; 199: 969–984.
Nakamura N, Oshiro N, Fukata Y, Amano M, Fukata M, Kuroda S et al. Phosphorylation of ERM proteins at filopodia induced by Cdc42. Genes Cells 2000; 5: 571–581.
Pietromonaco SF, Simons PC, Altman A, Elias L . Protein kinase C-theta phosphorylation of moesin in the actin-binding sequence. J Biol Chem 1998; 273: 7594–7603.
Gloerich M, ten Klooster JP, Vliem MJ, Koorman T, Zwartkruis FJ, Clevers H et al. Rap2A links intestinal cell polarity to brush border formation. Nat Cell Biol 2012; 14: 793–801.
ten Klooster JP, Jansen M, Yuan J, Oorschot V, Begthel H, Di Giacomo V et al. Mst4 and Ezrin induce brush borders downstream of the Lkb1/Strad/Mo25 polarization complex. Dev Cell 2009; 16: 551–562.
Belkina NV, Liu Y, Hao JJ, Karasuyama H, Shaw S . LOK is a major ERM kinase in resting lymphocytes and regulates cytoskeletal rearrangement through ERM phosphorylation. Proc Natl Acad Sci USA 2009; 106: 4707–4712.
He J, Lau AG, Yaffe MB, Hall RA . Phosphorylation and cell cycle-dependent regulation of Na+/H+ exchanger regulatory factor-1 by Cdc2 kinase. J Biol Chem 2001; 276: 41559–41565.
Lau AG, Hall RA . Oligomerization of NHERF-1 and NHERF-2 PDZ domains: differential regulation by association with receptor carboxyl-termini and by phosphorylation. Biochemistry 2001; 40: 8572–8580.
Weinman EJ, Steplock D, Zhang Y, Biswas R, Bloch RJ, Shenolikar S . Cooperativity between the phosphorylation of Thr95 and Ser77 of NHERF-1 in the hormonal regulation of renal phosphate transport. J Biol Chem 2010; 285: 25134–25138.
Voltz JW, Brush M, Sikes S, Steplock D, Weinman EJ, Shenolikar S . Phosphorylation of PDZ1 domain attenuates NHERF-1 binding to cellular targets. J Biol Chem 2007; 282: 33879–33887.
Fouassier L, Nichols MT, Gidey E, McWilliams RR, Robin H, Finnigan C et al. Protein kinase C regulates the phosphorylation and oligomerization of ERM binding phosphoprotein 50. Exp Cell Res 2005; 306: 264–273.
Shenolikar S, Voltz JW, Cunningham R, Weinman EJ . Regulation of ion transport by the NHERF family of PDZ proteins. Physiology 2004; 19: 362–369.
Terawaki S, Maesaki R, Hakoshima T . Structural basis for NHERF recognition by ERM proteins. Structure 2006; 14: 777–789.
Garbett D, Bretscher A . PDZ interactions regulate rapid turnover of the scaffolding protein EBP50 in microvilli. J Cell Biol 2012; 198: 195–203.
Garbett D, Sauvanet C, Viswanatha R, Bretscher A . The tails of apical scaffolding proteins EBP50 and E3KARP regulate their localization and dynamics. Mol Biol Cell 2013; 24: 3381–3392.
Mullin JM . Epithelial barriers, compartmentation, and cancer. Sci STKE 2004; 2004: pe2.
Shibata T, Chuma M, Kokubu A, Sakamoto M, Hirohashi S . EBP50, a beta-catenin-associating protein, enhances Wnt signaling and is over-expressed in hepatocellular carcinoma. Hepatology 2003; 38: 178–186.
Georgescu MM, Morales FC, Molina JR, Hayashi Y . Roles of NHERF1/EBP50 in cancer. Curr Mol Med 2008; 8: 459–468.
McClatchey AI . Merlin and ERM proteins: unappreciated roles in cancer development? Nat Rev Cancer 2003; 3: 877–883.
Clucas J, Valderrama F . ERM proteins in cancer progression. J Cell Sci 2015; 128: 1253.
Bruce B, Khanna G, Ren L, Landberg G, Jirstrom K, Powell C et al. Expression of the cytoskeleton linker protein ezrin in human cancers. Clin Exp Metastasis 2007; 24: 69–78.
Sarrio D, Rodriguez-Pinilla SM, Dotor A, Calero F, Hardisson D, Palacios J . Abnormal ezrin localization is associated with clinicopathological features in invasive breast carcinomas. Breast Cancer Res Treat 2006; 98: 71–79.
Li Q, Gao H, Xu H, Wang X, Pan Y, Hao F et al. Expression of ezrin correlates with malignant phenotype of lung cancer, and in vitro knockdown of ezrin reverses the aggressive biological behavior of lung cancer cells. Tumour Biol 2012; 33: 1493–1504.
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ . Cancer statistics. 2009 CA Cancer J Clin 2009; 59: 225–249.
Lengyel E . Ovarian cancer development and metastasis. Am J Pathol 2010; 177: 1053–1064.
Jesionowska A, Cecerska-Heryc E, Matoszka N, Dolegowska B . Lysophosphatidic acid signaling in ovarian cancer. J Recept Signal Transduct Res 2015; 35: 578–584.
Murph MM, Nguyen GH, Radhakrishna H, Mills GB . Sharpening the edges of understanding the structure/function of the LPA1 receptor: expression in cancer and mechanisms of regulation. Biochim Biophys Acta 2008; 1781: 547–557.
Lengyel E, Burdette JE, Kenny HA, Matei D, Pilrose J, Haluska P et al. Epithelial ovarian cancer experimental models. Oncogene 2014; 33: 3619–3633.
Acknowledgements
We are very grateful to Dr Monique Arpin (CNRS/Institut Curie, France) for the Ezrin WT and T567 mutant constructs, and also to Dr Andrew Godwin (Fox Chase Cancer Center, PA) for the HIO-80 cell line. We thank the members of our laboratories for their contributions and helpful discussions. This work was supported by the DGIST MIREBraiN R&D program (16-BD-0402 to YSO), Basic Science Research Program (2016010015, 2014R1A1A1003159 to YSO), the Korea Brain Research Institute basic research program (2231-415 to YSO) by the Ministry of Science, ICT & Future Planning of Korea, and the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2016R1D1A1B03935615 to YSO, 2016R1A6A3A11934335 to JJH). In addition, the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (no 2010-0028684 to PGS) supported this work.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Experimental & Molecular Medicine website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Oh, YS., Heo, K., Kim, EK. et al. Dynamic relocalization of NHERF1 mediates chemotactic migration of ovarian cancer cells toward lysophosphatidic acid stimulation. Exp Mol Med 49, e351 (2017). https://doi.org/10.1038/emm.2017.88
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2017.88
This article is cited by
-
Role of lysophosphatidic acid and its receptors in health and disease: novel therapeutic strategies
Signal Transduction and Targeted Therapy (2021)
-
Polarity scaffolds signaling in epithelial cell permeability
Inflammation Research (2021)

{kind=link}