
Abstract
We evaluated the role of IL-10- in IL-33-mediated cholesterol reduction in macrophage-derived foam cells (MFCs) and the mechanism by which IL-33 upregulates IL-10. Serum IL-33 and IL-10 levels in coronary artery disease patients were measured. The effects of IL-33 on intra-MFC cholesterol level, IL-10, ABCA1 and CD36 expression, ERK 1/2, Sp1, STAT3 and STAT4 activation, and IL-10 promoter activity were determined. Core sequences were identified using bioinformatic analysis and site-specific mutagenesis. The serum IL-33 levels positively correlated with those of IL-10. IL-33 decreased cellular cholesterol level and upregulated IL-10 and ABCA1 but had no effect on CD36 expression. siRNA-IL-10 partially abolished cellular cholesterol reduction and ABCA1 elevation by IL-33 but did not reverse the decreased CD36 levels. IL-33 increased IL-10 mRNA production but had little effect on its stability. IL-33 induced ERK 1/2 phosphorylation and increased the luciferase expression driven by the IL-10 promoter, with the highest extent within the −2000 to −1752 bp segment of the 5′-flank of the transcription start site; these effects were counteracted by U0126. IL-33 activated Sp1, STAT3 and STAT4, but only the STAT3 binding site was predicted in the above segment. Site-directed mutagenesis of the predicted STAT3-binding sites (CTGCTTCCTGGCAGCAGAA→CTGCCTGGCAGCAGAA) reduced luciferase activity, and a STAT3 inhibitor blocked the regulatory effects of IL-33 on IL-10 expression. Chromatin immunoprecipitation (CHIP) confirmed the STAT3-binding sequences within the −1997 to −1700 and −1091 to −811 bp locus regions. IL-33 increased IL-10 expression in MFCs via activating ERK 1/2 and STAT3, which subsequently promoted IL-10 transcription and thus contributed to the beneficial effects of IL-33 on MFCs.
Similar content being viewed by others


Introduction
IL-33, a recently discovered cytokine that belongs to the IL-1 superfamily, activates many types of cells and induces Th2 cytokines via its transmembrane receptor, ST2L.1 IL-33 has both harmful and beneficial effects, depending on the circumstances and pathological processes involved.2 This cytokine mediates a number of immunology disorders (for example, anaphylactic reactions, systemic sclerosis and systemic lupus erythematosus).2
However, IL-33 exerts a wide range of protective effects in the cardiovascular system. For example, IL-33 promotes anti-oxidized low-density lipoprotein (ox-LDL) antibody production and greatly attenuates atherosclerosis in vivo.3 Moreover, an in vitro study demonstrated that IL-33 protects lipid-overloading in macrophage-derived foam cells (MFCs) via regulating the expression of genes involved in cholesterol transport, such as CD36 and ATP-binding cassette transporter A-1 (ABCA1).4 IL-10 is an important cytokine in regulating cholesterol transport and has been shown to prevent macrophages from cholesterol overload and to stimulate cholesterol efflux.5, 6 In addition, IL-10 also increases ABCA1 expression.7, 8
The associations between IL-33 and IL-10 are unknown, and studies have reported conflicting results. Several studies have shown that the IL-10 levels were unchanged by IL-33 treatment, whereas others have reported the opposite outcomes, depending on the different physio-pathological processes evaluated and the animal models utilized.3, 9 Furthermore, studies have shown that IL-33 directly induced CD4+CD25+Foxp3+ regulatory T cells in vivo,10 which may also support a role for IL-33 in promoting IL-10 production, as IL-10 is thought to be abundantly expressed in these cells.11
Therefore, we hypothesized that IL-33 promotes IL-10 production in MFCs, which helps to regulate cholesterol transport-related genes, which contribute to the beneficial effects of IL-33 therapy in MFCs. We first examined whether there was a link between IL-33 and IL-10 in coronary artery disease (CAD) patients. Thereafter, an in vitro study was performed to validate the association between IL-33 and IL-10 and the role that IL-10 plays in the reduction of the intracellular cholesterol levels in MFCs by IL-33. Moreover, we investigated whether the promotion of IL-10 expression by IL-33 in macrophages was transcription-related and identified related transcription factors and potential binding sites.
Materials and methods
Patients
A total of 229 angiographically confirmed stable CAD patients were enrolled in the Department of Cardiology, Sun Yat-sen Memorial Hospital from January to December 2011. The following major exclusion criteria were applied: (1) heart failure; (2) myocarditis; and (3) allergic diseases (that is, asthma), autoimmune diseases (that is, systemic lupus erythematous), inflammatory diseases (for example, rheumatic arthritis and inflammatory bowel disease) and malignant diseases. All participants were Han Chinese. The study protocol was approved by the ethics committee of Sun Yat-sen Memorial Hospital, which is affiliated with Sun Yat-sen University, and written consent was obtained from each participant (or a relative).
Cell culture, site-directed mutagenesis, vector construction and transfection of siRNAs and plasmids
The human monocyte cell line THP-1 was purchased from ATCC (Bethesda, MD, USA). The cells were plated at a density of 5 × 105 ml−1 and incubated in 1640 medium (Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum (Invitrogen) containing 1% penicillin (Invitrogen), 1% streptomycin (Invitrogen), 2 mM L-glutamine (Invitrogen) and 50 μM 2-mercaptoethanol (Sigma, Saint Louis, MO, USA) at 37 °C and 5% CO2. The cells were differentiated into macrophages using 160 nM phorbol 12-myristate 13-acetate (PMA) (Sigma) intervention for 72 h. The macrophages were treated with ox-LDL-c (50 μg ml−1) and later stimulated with different doses of IL-33 (1, 10, 30 or 100 ng ml−1, Sino Biological, Beijing, China) for various durations. Phosphate-buffered saline (PBS)/0.1% bovine serum albumin was used as vehicle control for in vitro human IL-33 stimulation experiments.
For siRNA transfection, stealth siRNA specific to the 3′-UTR of the IL-10 mRNA was used as previously described.12 Details of the sequence are listed in the Supplementary Materials. For transfection, siRNA (50 nM) was transfected into MFCs using Lipofectin 2000 (Invitrogen) as per the manufacturer’s instructions. A nonsense scramble siRNA was used as a transfection control (Supplementary Materials). The knockdown efficiency was tested by quantitative real-time PCR (qRT-PCR) and ELISA for each experiment. All siRNAs were provided by the Ribobio Company (Guangzhou, Guangdong, China).
To identify the core sequences of the IL-10 promoter targeted by IL-33, a promoter deletion analysis was performed. Briefly, a total of nine segments were amplified by PCR, which were later ligated to the pGL3-Basic vector using the incision enzymes KpnI and XhoI to construct pGL3-Basic-IL 10-1 to pGL3-Basic-IL10-9. Briefly, of the two to nine inserts, ~250 bp in the 5′-flank were gradually deleted; therefore, vectors containing decremented sequences of the 5′-flank of the IL-10 gene were constructed. The plasmids (pGL3-Basic, pGL3-Basic-IL10-1 to pGL3-Basic-IL10-9) and inner-control plasmid (pRL-TK) were co-transfected into MFCs using Lipofectamine LTX & PLUS (Invitrogen) for luciferase activity assessment as described below.
A core sequence of the STAT3-binding site (TTCC) in the potential target segments were deleted (TTCC→C) by site-directed mutagenesis using a commercially available kit (Stratagene, La Jolla, CA, USA). Briefly, mutated plasmids were obtained by PCR using site-directed mutagenesis primers (listed in Supplementary Materials) and wild-type plasmid as templates. DpnI was used to digest the remaining wild-type plasmid, and PCR products were transformed into the provided XL1-Blue competent cells. Finally, mutated plasmids were extracted and subsequently transfected as above.
Biochemical analyses
ELISA
Blood samples were obtained at admission, and serum was collected by centrifugation, which was frozen immediately at −80 °C for subsequent batch analyses. Serum levels of IL-33 and IL-10 were measured using ELISA kits (IL-33: Enzo, Farmingdale, NY, USA; IL-10: R&D Systems, Minneapolis, MN, USA). Cell supernatants following the treatments were collected and were subjected to IL-10 determinations after centrifugation.
Total cellular cholesterol assessments
Cellular cholesterol levels were analyzed 48 h after stimulation or 72 h after siRNA transfection, as previously described.13 Briefly, MFCs were washed with PBS and subsequently lysed. An aliquot (20 μl) was used for total protein assessment, and the rest of the cell lysates were used for lipid extraction using a mixture of hexane and isopropanol (3:2, v/v), as described previously.14 The intracellular cholesterol content was assessed using a commercially available kit (BioAssay System, Hayward, CA, USA).
Quantitative real-time RT-PCR
Reverse transcription polymerase chain reaction (RT-PCR) was performed as previously described.15 Briefly, total RNA was extracted using Trizol (Invitrogen) and was used for reverse transcription. qRT-PCR was performed using the primers listed in the Supplementary Materials.
Immunofluorescence and flow cytometry
Flow cytometry and immunofluorescence were performed to demonstrate the presence of ST2L on MFCs. For immunofluorescence, treated cells were washed and fixed in 4% paraformaldehyde. Cell membranes were permeabilized by 1% Triton. Rabbit-anti human CD68 (Cell Signaling Technology, Danvers, MA, USA) and goat-anti human ST2L (R&D Systems) primary antibodies were added followed by mouse anti-rabbit IgG Alexa 488-labeled and mouse anti-goat Cy3-labeled secondary antibodies. Finally, cell nuclei were re-stained with DAPI and examined by confocal fluorescence microscopy (LSM710, Carl Zeiss, Oberkochen, Germany).
For flow cytometry, MFCs were collected and washed. Cell precipitations were fixed and permeabilized using a Fix & Perm kit (Life Technologies, Bedford, MA, USA). Goat-anti human ST2L (R&D Systems) and rabbit anti-goat IgG FITC-labeled antibodies (Jackson ImmunoResearch, West Grove, PA, USA) were added. After that step, the cells were washed and incubated with Alexa Fluor 647-labeled anti-CD68 (Abcam, EPR1392Y, Cambridge, MA, USA). Finally, the cells were collected, resuspended in PBS and analyzed by flow cytometry. Autofluorescence and isotype controls were run routinely as controls.
Western blot analysis
Western blot analysis was conducted as described previously.15 Briefly, total protein (for Sp1, nuclear protein was extracted according to the manufacturer’s instructions, Promega, Madison, WI, USA) was extracted using RIPA (with PMSF) and was analyzed by SDS-PAGE, which was blotted onto polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). The membranes were blocked and incubated overnight with the indicated primary and secondary antibodies (primary antibodies: CD36, Novus, Littleton, CO, USA; ABCA1, Abcam; and GAPDH, ERK, p38, JNK, Sp1, Histone H3, STAT3 and STAT4, Cell Signaling Technology; secondary antibody: HRP-conjugated anti-mouse/rabbit IgG (Jackson ImmunoResearch)). Visualization of immunoreactive human IL-33 was performed using enhanced ECL reagents (Thermo Fisher Scientific, Rockford, IL, USA). For the detection of ST2L presence in the MFC membrane, total membrane proteins were extracted using a CelLytic MEM protein extraction kit (Sigma), and were subsequently subjected to routine western blot analysis, as described above, and anti-ATPase Na+/K+-β2, instead of GAPDH, antibody (Abcam) was used as a loading control.
IL-10 mRNA stability detection and dual-luciferase reporter gene assays
IL-10 mRNA is unstable; therefore, stabilization of its mRNA is important for its expression.16 To test whether IL-33 stabilizes IL-10 mRNA, we tested the half-life of IL-10 mRNA as previously described.17 Briefly, MFCs were treated with different doses of IL-33 for 6 h, and then actinomycin D (ActD, 5 μg ml−1) was added to block RNA synthesis. The remaining IL-10 mRNA levels were measured using qRT-PCR, as described above.
It is widely accepted that transcriptional regulation and promoter activity is crucial to IL-10 expression.11 To assess directly the effects of IL-33 on IL-10 promoter activity, we performed dual-luciferase reporter assays using a previously reported method but with modifications.18 Briefly, re-constructed pGL3 and pRL-TK vectors were transfected into MFCs, as described above, the cells were collected and luciferase activity was determined using a luminometer and a commercial kit (Promega).
Bioinformatic analysis of IL-10 promoter sequences
To explore the potential transcriptional factors that mediate the upregulatory effects of IL-33 on IL-10 mRNA production, we used a published formula, namely, the Genomatrix MatInspector (Transcription factor binding sites, weight matrices; Matrix Library version 9.4; Core similarity at 0.75 and Matrix similarity at ‘Optimized’), to predict potential transcription factor binding sites. To include more possible target sequences, we expanded to the −2000 bp of the 5′-flank of the IL-10 gene sequence from transcription start sites instead of the −1531 bp, as previously reported.19 The predictive results from the Genomatrix MatInspector were confirmed using the JASPAR database to provide further support of the results. Potential homologous sequences to the predicted binding sequence in different species were identified using a UCSC database and homology analyses were conducted using Clustal X2.
Chromatin immunoprecipitation
To validate that STAT3 binds the target sequences predicted by the bioinformatic analysis, we performed chromatin immunoprecipitation using a Pierce Agarose ChIP Kit (Thermo Fisher Scientific). Briefly, cells were cross-linked in 1% formaldehyde followed by sequential cell and nuclear membrane lysis. After the second lysis (nuclear lysis), sonication was performed to generate sheared DNA fragments. Supernatants of the final lysates were transferred to an immunoprecipitation system containing a spin column and rabbit anti-human STAT3 antibody (Cell Signaling Technology). The DNA–protein–STAT3 antibody complex was precipitated with protein A/G plus agarose for later elution. After elution, DNA was recovered and extracted for qPCR detection (primers specific to potential target sequences (listed in the Supplementary Materials) were identified by promoter deletion and bioinformatic analyses). Routine input control using 10% volume of the sonicated lysate was performed.
Statistical analysis
Normally distributed data are expressed as the mean±s.d.; otherwise, they are reported as the median and 25th and 75th quartiles. Categorical variables are presented as absolute and relative frequencies. Spearman correlations were used to evaluate the association between IL-33 and IL-10 serum levels. Statistical comparisons across groups were made using t-tests (or one-way ANOVA followed by SNK test for multiple post hoc comparisons if three or more groups were involved) for normally distributed data, and the Pearson’s χ2-test was used for categorical variables comparisons. For non-normally distributed data, the Wilcoxon two-sample test was used. All statistical analyses were performed using SAS software version 9.13 (Cary, NC, USA), and statistical graphs were produced using GraphPad Prism Software (San Diego, CA, USA).
Results
Patients’ characteristics and IL-33 and IL-10 levels
A total of 229 CAD patients (54 with acute myocardial infarction and 175 with stable angina pectoris) were included. The basic characteristics of the two groups are listed in Table 1. Overall, age, gender, smoking habit, blood pressure and medications were not significantly different among the groups. The serum IL-33 levels in the patients were not normally distributed, whereby the minimum, P25, median, P75 and maximum levels were 0.14, 24.25, 56.67, 259.52 and 946.18 pg ml−1, respectively (Figure 1a). Similarly, the IL-10 concentrations were not normally distributed, although to a considerably lesser extent (the minimum, P25, median, P75 and maximum levels were 0.64, 10.48, 18.85, 23.46 and 45.71 ng ml−1, respectively, Figure 1a). To analyze the relationship between IL-33 and IL-10, patients were divided into the following two groups according to their median serum IL-33 levels: low IL-33 level group (L-group, all IL-33 levels⩽56.67 pg ml−1, n=115) and high IL-33 level group (H-group, all IL-33 levels>56.67 pg ml−1, n=114). The minimum, P25, median, P75 and maximum levels were 0.64, 7.60, 12.92, 18.49 and 45.71 ng ml−1, respectively, for the L-group and were 1.10, 19.67, 27.31 and 42.68 ng ml−1, respectively, for the H-groups, with significant differences observed (Z=6.87, P<0.01; Figure 1b). Furthermore, a Spearman correlation analysis yielded a positive association between the serum IL-33 and IL-10 concentrations, with a correlation coefficient of 0.503 (Figure 1c). In addition, the LDL-c and TC levels were also lower in the L-group patients, whereas the HDL-c and TG levels did not differ between the groups (Table 1). A logistic regression analysis showed that such differences were still significant after adjusting for serum creatinine, TC, LDL-c, HDL-c and hs-CRP (Wald χ2=15.00, P<0.01).

Serum levels of IL-33 and IL-10 and their relationship in coronary artery patients. (a) Serum levels of IL-33 and IL-10 in the included patients. (b) Serum levels of IL-10 were lower in those with low serum IL-33 concentrations. (c) Serum levels of IL-10 positively correlated with those of IL-33.
The IL-33 membrane receptor ST2L is constitutively expressed in MFCs
The transmembrane G-protein-coupled receptor ST2L is the only recognized receptor of IL-33 and is believed to mediate most of the biological effects of IL-33. Therefore, we first examined the presence of this receptor on the surface of MFCs. Moreover, as the presence of ST2L mRNA has been documented previously, we confirmed the presence of this receptor at the protein level.
Immunofluorescence showed substantial ST2L expression, which was as abundant as CD68 expression in the MFCs (Figure 2a–d). To calculate the proportion of CD68+/ST2L+ MFCs, we performed flow cytometry. As shown in Figure 2e, 95.7% and 98.21% of the MFCs were CD68 and ST2L positive, respectively. Meanwhile, 94.24% of the MFCs were positive for both CD68 and ST2L (Figure 2e). This finding indicated that ST2L was constitutively expressed on the surface of the MFCs. However, the expression profile of ST2L on the surface of the membrane could not be identified; because we permeabilized the cell membrane the location of the segment that the antibody recognized was unclear. Moreover, it may be difficult for the antibody to discriminate ST2L from the soluble form. Therefore, we extracted membrane protein and examined ST2L expression by western blotting. As shown in Figure 2f, ST2L expression could be detected in the MFC membrane. These data consistently demonstrate the constitutive presence of ST2L on the surface of MFCs.

Validation of the presence of ST2L on MFC membrances. (a–d) Immunofluorescence staining of DAPI, CD68 and/or ST2L. (e) Flow cytometry measurements of the proportions of CD68+ST2L+ cells. (f) Western blot analysis of cell membrane proteins. upper lane, ST2L; lower lane, ATPase Na+/K+-β2. The data in a–d represent two independent experiments and the data in e, f represent three independent experiments. MFC, macrophage-derived foam cell.
Moderate IL-33 stimulation induced IL-10 expression in MFCs
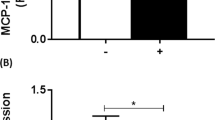
To validate the effects of IL-33 on IL-10 production, we assessed IL-10 expression in MFCs using different doses of IL-33. Results from real-time RT-PCR showed that IL-10 was upregulated by IL-33 in a dose-dependent manner, with concentrations ranging from 1 ng ml−1 to a peak of 30 ng ml−1, which was more than five times higher than the PBS-treated samples (Figure 3a). Interestingly, when stimulated with larger doses (50 and 100 ng ml−1), the IL-10 mRNA abundance was decreased compared with that of the 30 ng ml−1 dose (Figure 3a). In addition, IL-10 mRNA determinations at different durations of IL-33 treatment showed that IL-33 regulated IL-10 mRNA expression profiles in a rapid manner, reaching a peak at 6 h after IL-33 intervention (Figure 3a). The IL-10 content in the cultured medium showed similar results (Figure 3b). These results showed that IL-10 was upregulated by IL-33 in MFCs under moderate to high concentration stimulus.

IL-33 upregulated IL-10 expression in MFCs in a dose-dependent manner within moderate concentrations. (a) The IL-10 mRNA abundance was increased upon IL-33 stimulation. (b) IL-10 supernatant levels from MFCs were upregulated following IL-33 treatment for 24 h. *P<0.05 versus baseline (PBS treatment); #P<0.05 versus high IL-33 concentrations (100 ng ml−1) treatment. These data represent three independent experiments. MFC, macrophage-derived foam cell.
IL-10 was involved in IL-33-induced cholesterol reduction and increased ABCA1 expression in MFCs
The above data showed that IL-33 robustly promoted IL-10 production in MFCs. IL-33 was shown to reduce cellular cholesterol, downregulate CD36 expression and increase the ABCA1 levels. Meanwhile, IL-10 was also shown to potently upregulate ABCA1 expression and increased cholesterol efflux, resulting in a reduction of total cellular cholesterol, which was similar to the effects of IL-33. To assess the IL-33 induction of IL-10 and the role that IL-33 plays in lipid-overload attenuation and alterations of CD36 and ABCA1 expression, we used siRNA against IL-10 to knockdown its expression. Under IL-33 stimulation, the siRNA-IL10 at a concentration of 75 nM reduced IL-10 mRNA to ~17% of the scramble siRNA level, resulting in a similar IL-10 mRNA abundance to the unstimulated MFCs (data not shown).
Cellular cholesterol levels, and CD36 and ABCA1 expression levels, under various treatments were determined under IL-33 stimulus with or without siRNA against IL-10. As expected, the IL-33 treatment resulted in more than 50% reduction in the total cellular cholesterol level (Figure 4a). IL-10 siRNA transfection significantly neutralized the IL-33-mediated lipid reduction in the MFCs, whereby it increased the cellular cholesterol contents by 1.86-fold compared with IL-33 alone and by 1.55-fold compared with IL-33 plus scramble siRNA treatment (Figure 4a). Compared with IL-33 alone, transfection with the same concentration of scramble siRNA slightly increased the cellular cholesterol level but this result was not statistical significant (Figure 4a).

Effects of IL-10 siRNA on IL-33-induced cellular cholesterol reduction and alterations of CD36 and ABCA1. (a) IL-33 reduced the cellular cholesterol content, which was abolished by IL-10 siRNA. (b, d, e) IL-33 downregulated whereas IL-10 siRNA had no effect on CD36 expression. (c, d, f) IL-33 increased IL-10 expression, which was diminished by siRNA against IL-10. *P<0.05 versus baseline (PBS treatment). These data represent three independent experiments. si-IL-10, siRNA against IL-10; si-Scr, siRNA with scramble sequences without any known target mRNA; TC, total cholesterol.
Consistent with the above findings, IL-33 treatment resulted in a 29% reduction in CD36 mRNA and a 38% reduction at the protein level (Figure 4b). Notably, IL-10 siRNA not only reversed the CD36 reduction but also further decreased CD36 levels. IL-33 increased ABCA1 expression, with a nearly two-fold increase in mRNA abundance and a more than two-fold increase at the protein level (Figure 4c). Importantly, IL-10 siRNA significantly reversed the ABCA1 upregulation by IL-33, with a nearly 35% reduction at the protein levels (Figure 4c). Transfection with scramble siRNA did not affect the regulatory effects of IL-33 on both CD36 and ABCA1 (Figure 4b–f). Taken together, these data indicate that IL-10 mediates IL-33-induced cholesterol reduction and increases in ABCA1 in MFCs.
ERK 1/2 activation in IL-33-induced IL-10 gene expression
It has been suggested that MAPKs are important for IL-10 synthesis in macrophages.20, 21 Recently, a study showed that IL-33 activates MAPKs in various cells;22 however, the relationship between MAPK activation and IL-33-induced IL-10 production has not been explored. To determine whether the ERK 1/2 signal mechanism is responsible for the upregulation of IL-10 by IL-33 in MFCs, we examined the activation states of ERK 1/2 in MFCs upon IL-33 stimulation. Treatment with IL-33 led to a slow but prolonged phosphorylation of ERK 1/2 (Figure 5a), reaching a peak at 30–45 min. The IL-33-mediated increase in IL-10 production was blocked by U0126 (Figure 5b and c), a specific inhibitor of ERK 1/2. The other MAPKs, p38 and JNK, have been demonstrated to play a crucial role in LPS-induced and anandamide-induced IL-10 production;18 however, the inhibition of these MAPKs had no effect on IL-10 production, although IL-33 also induced their phosphorylation (data not shown). These data indicate that ERK 1/2, rather than p38 and JNK, plays an important role in IL-33-induced IL-10 production.

Effects of IL-33 on ERK 1/2 phosphorylation and its role in IL-33-induced IL-10 expression. (a) IL-33 induced ERK 1/2 phosphorylation; (b, c) an ERK 1/2 inhibitor reversed IL-10 upregulation by IL-33. These data represent three independent experiments. p-ERK 1/2, phosphorylated ERK 1/2; t-ERK 1/2, total ERK 1/2.
IL-33 activated the IL-10 promoter but did not increase IL-10 mRNA stability
Previous studies have shown that both transcriptional and post-transcriptional mechanisms are involved in the regulation of IL-10 expression. To explore the two mechanisms in IL-33-induced IL-10 production, we used the transcription inhibitor actinomycin D (ActD, 5 μg ml−1) to stop mRNA synthesis and detect changes in IL-10 mRNA levels.
Treatment with IL-33 (30 ng ml−1) for 6 h resulted in increased IL-10 mRNA, which could be completely blocked by ActD (Figure 6a). This finding suggested that the induction of IL-10 by IL-33 was transcription dependent. To detect whether IL-33 stabilizes IL-10 mRNA, we pre-incubated MFCs with IL-33 for 6 h; next, ActD was added, and the remaining IL-10 mRNA levels at the indicated times were determined. As shown in Figure 6b, the remaining IL-10 mRNA did not differ significantly between the IL-33 and PBS treatments at any of the time points indicated. These results indicated that IL-33 did not alter IL-10 mRNA stability.

Effects of IL-33 on IL-10 mRNA production, stability and gene promoter activity. (a) A transcription factor inhibitor, ActD, abolished the up-regulatory effects of IL-10 in MFCs. (b) IL-33 had a scarcely detectable influence on IL-10 mRNA stability. (c) IL-33 activated IL-10 gene promoter, as indicated by luciferase activity. (d) Promoter deletion analysis, which identified core sequences in the IL-33-induced IL-10 production process. *P<0.05 versus PBS treatment; #P<0.05 versus the pGL3-Basic. These data represent three independent experiments. ActD, actinomycin D; MFC, macrophage-derived foam cell.
To assess directly whether IL-33 activates the IL-10 promoter, we performed dual-luciferase reporter gene assays. As shown in Figure 6c, compared with pGL3-Basic, the promoter containing the 5′-flank −2000 bp sequence of the IL-10 gene was greatly activated by IL-33, with ~30-fold luciferase activity induction compared with PBS-treated cells. The ERK 1/2 inhibitor U0126 significantly reduced the luciferase activity upregulated by IL-33 (Figure 6c). These data indicated that IL-33 directly activated the IL-10 gene promoter via ERK 1/2.
It is unlikely that IL-33 activates the entire −2000 bp 5′-flank of the IL-10 gene, which includes the transcription start site; therefore, we next explored the core sequence that is targeted by IL-33. As shown in Figure 6d, compared with the pGL3-Basic-IL10-2 to pGL3-Basic-IL10-9 vectors, which did not contain the −2000 to −1752 bp 5′-flank sequence, pGL3-Basic-IL10-1 exhibited the maximal upregulated promoter activity response to IL-33, as indicated by luciferase activity assessments, followed by pGL3-Basic-IL10-4, pGL3-Basic-IL10-5, pGL3-Basic-IL10-7, and pGL3-Basic-IL10-9, with approximately 70.3%, 70.8% and 22.3% luciferase activity of the pGL3-Basic IL10-1 group, respectively. These data demonstrated that the target sequences that are targeted by IL-33 are probably within the −2000 to −1752 bp region; however, whether the sequences located within the −1080 bp have potential roles cannot be ruled out.
STAT3 as a transcription factor in the regulatory process of IL-33-mediated IL-10 expression
Transcriptional activity is largely determined by transcription factors, but it is unclear which transcription factors might be involved in IL-33-induced IL-10 production. Previous studies have reported that Sp1, STAT3 and STAT4 mediate ERK 1/2 kinase-dependent activation of the IL-10 transcriptional process.20, 23, 24, 25 Therefore, we examined phosphorylation of Sp1, STAT3 and STAT4 upon IL-33 intervention. As shown in Figure 7a, IL-33 induced phosphorylation of all three transcription factors, with the greatest alterations occurring in STAT4 followed by Sp1 and STAT3.

Identification of transcription and target sequences that react with IL-33. (a) Effects of IL-33 on intra-nuclear Sp1 expression as well as STAT3 and STAT4 phosphorylation. (b) Bioinformatic analysis of STAT3-binding sites within the −2000 to −1752 bp of the 5′-flank of the IL-10 transcription start site. (c) Homology analysis of the predicted binding STAT3 site among five mammalian species. (d) Effects of TTC base deletion by site-directed mutagenesis on promoter activity indicated by luciferase activity. (e, f) Effects of a STAT3 inhibitor, cryptotanshinone, on IL-33-induced IL-10 mRNA (e) and protein (f) production. (g) CHIP analysis using a STAT3 antibody and primers specific to IL-10 promoter segments following PBS or IL-33 (30 ng ml−1) treatment. p-STAT3, phosphorylated STAT3; p-STAT4, phosphorylated STAT4; t-STAT3, total STAT3; t-STAT4, total STAT4; wild type, pGL3-Basic-IL10.1-Wild type; Mutant, pGL3-Basic-IL10.1-Mutant; IL-33-P1, qPCR using primer set 1; IL-33-P4, qPCR using primer set 6 (all primer sets are listed in Supplementary Materials). *P<0.05 versus PBS treatment; #P<0.05 versus IL-33-P1. The data in a and d–g represent three independent experiments.
It is unknown which of the above transcription factors influence IL-10 expression and a lack of a STAT4 inhibitor made it difficult to make a clear discrimination. Therefore, we performed a bioinformatic analysis of the IL-10 promoter sequences to predict which may serve a role. The −2000 bp at the 5′-flank of the IL-10 gene sequence from the transcription start site (chromosome: hg38 chr1:206766943 to 206768942) was retrieved using the UCSC genome browser. Details of the sequence and results from the Genomatix MatInspector prediction are listed in Supplementary Materials. Briefly, a total of 602 transcription factor binding sites were identified, and 508 of them had a matrix similarity above 0.8. Because the −2000 to −1752 bp fragment had the highest extent of increased luciferase activity, we focused on these sequences and identified 58 potential transcription factors and binding sites (Supplementary Materials). Notably, STAT3 was predicted to bind −1954 to −1936 bp (5′-CTGCTTCCTGGCAGCAGAA-3′, Figure 7b) of the forward strand with a matrix similarity as high as 0.961 by the Genomatrix Matlnspector. Interestingly and most importantly, the results from the JASPAR database showed an identical sequence in both forward and reverse chains at the same location, with scores of 12.48 and 5.45, respectively (forward chain: −1951 to −1941 bp, 5′-CTTCCTGGCAG-3′; reverse chain: 5′-CTGCCAGGAAG-3′, Figure 7b). Homology analysis showed that the sequence is highly conserved, with an identity of 84.67% (Figure 7c) among different mammalian species, indicating a functional region contained in this sequence. To validate this conservation, we deleted the TTC bases to disrupt the predicted binding site of STAT3 (CTGCTTCCTGGCAGCAGAA→CTGCCTGGCAGCAGAA) and the results from the luciferase activity assessment supported that the mutant significantly reversed the transcriptional activity induced by IL-33, with a reduction in luciferase activity exceeding 50% (Figure 7d). In addition, a specific STAT3 inhibitor, cryptotanshinone, was administered and the IL-10 expression levels were determined. As shown in Figure 7e and f, cryptotanshinone reversed, at least in part, the upregulation of IL-10 by IL-33. All of these results show that IL-33 phosphorylates STAT3, which facilitates its binding to target sequences (−1954 to −1936 bp may be involved) on the IL-10 promoter and promotes transcription.
To validate these expected results, we performed a CHIP analysis in MFCs stimulated with IL-33 using different sets of IL-10 specific promoters spanning distinct potential target sequences. The CHIP analysis showed that STAT3 was mobilized mostly to the region containing −1997 to −1700 bp and −1091 to −811 bp, up to ~1.7-fold compared with other regions in the IL-10 promoter and 2.9-fold compared with PBS-treated samples (Figure 7g). These data confirm the above predictions but did not exclude the interaction of STAT3 with other sequences in the promoter region that are considerably closer to the transcription start site.
Discussion
Both IL-33 and IL-10 benefit atherosclerosis and reduce foam cell formation; however, their interactions in this process are unknown. We showed that the serum IL-33 and IL-10 levels were positively correlated in CAD patients, and those with higher serum IL-33 levels tended to have higher IL-10 concentrations. Our in vitro study demonstrated that IL-33 induced cholesterol reduction and ABCA1 expression, at least in part, via the upregulation of IL-10 in MFCs. However, IL-10 upregulation was not related to the IL-33-induced CD36 decrease. Furthermore, we showed that IL-33 activated ERK 1/2 and STAT3, which subsequently facilitated STAT3 to bind to the −1954 to −1936 bp sequence upstream of the IL-10 transcription start site, thereby promoting its transcription in MFCs.
Regulatory effects of IL-10 production in macrophages by IL-33
The regulation of IL-10 expression in immune cells is complex and involves a wide range of mechanisms.11 It is widely accepted that transcriptional regulation plays a pivotal role in IL-10 gene expression.11, 26 Indeed, ERK-dependent transcription activity mechanisms in IL-10 production have been demonstrated.27 A number of studies have reported that IL-33 activates MAPKs, but to the best of our knowledge, no study has explored the relationship between IL-33-induced MAPK activation and IL-10 expression. Conversely, the effects of IL-33 on IL-10 production remains unknown. Our study showed that IL-33 directly activates the IL-10 promoter, which was significantly neutralized by an ERK 1/2 inhibitor. This finding could help to elucidate the interactions among IL-33, the ERK 1/2 pathway and IL-10 production in MFCs.
In addition to classical transcriptional regulatory mechanisms, post-transcriptional regulation of IL-10 expression has also been reported.16, 28, 29 For example, microRNA-106a and microRNA-98 have been shown to negatively regulate IL-10 expression.28, 29 More importantly, the regulatory mechanisms of microRNA-106a have been discovered, and the results showed that microRNA-106a can also be enhanced by a common transcription factor, namely, Sp1. This finding may explain the inability to promote IL-10 production in MFCs when larger doses of IL-33 were used, as excessively activated Sp1 may lead to increased microRNA production,30, 31 which could result in degradation in IL-10 mRNA.29 However, the involved microRNAs are largely unknown, and more studies in this field should be encouraged.
IL-10 promoter sequences in Burkitt’s lymphoma have been reported previously.19 It was concluded that the sequence from −1318 to +27 bp from the transcriptional site of the IL-10 gene was the main regulatory region.19 However, this finding does not contradict our result showing regulatory target sequences detected far from this region because the current study did not exclude sequences outside this region that exert regulatory properties. Indeed, despite not as potent as the −2000 to −1752 bp sequences in promoting IL-10 transcription, the promoter deletion analysis revealed more regulatory regions within −1318 bp region (three segments, as stated in the results). Moreover, the CHIP analysis supported the role of the reported sequences19 in the regulation of STAT3–DNA interactions. Therefore, the current study identified the main regulatory elements that interact with STAT3-mediated IL-10 upregulation but did not exclude other sequences that may play a role. In keeping with this notion, the authors’ claimed that the AAAATT sequence located at position −2209 bp may also play a role in IL-10 transcription19 and it seems plausible that both the distal and proximal regions in the IL-10 promoter are important in transcriptional regulation.
IL-33, IL-10, lipid accumulation and cholesterol transporters in MFCs
ABCA1 and CD36 are the most important cholesterol transporters, and studies have shown that the regulation of these two proteins is the common beneficial mechanisms shared by both IL-33 and IL-10 in MFCs.4, 5 For example, IL-33 was shown to increase ABCA1 expression and macrophages sources of IL-10 were demonstrated to directly upregulate ABCA1.5 The mechanisms of IL-33-induced ABCA1 expression are largely unknown, and several researchers speculated that endogenous IL-33 acts as a transcription factor and directly promotes ABCA1 transcription.4 However, this function does not explain mechanisms of exogenous IL-33. Our in vitro study showed that siRNA against IL-10 greatly neutralized IL-33-induced cholesterol reduction and ABCA1 expression in MFCs, indicating that IL-10 could, at least in part, mediate IL-33-induced cholesterol efflux and ABCA1 expression. Considering the in vivo finding from CAD patients showing that low IL-33 levels were associated with low IL-10 concentrations in the serum, it is plausible to speculate that a IL-33/IL-10 axis may exist in CAD patients to regulate CAD development.
In contrast to ABCA1, CD36 does not seem to play an important role in the interactions among IL-33, IL-10 and cholesterol transport. Despite a study that showed that IL-33 reduced CD36 expression in MFCs,4 the role of IL-10 in CD36 expression is controversial. On the one hand, a previous study has shown that IL-10 downregulated CD36.5 On the other hand, a recent study showed that IL-10 has no effect on ox-LDL-induced CD36 expression6 and some even showed that IL-10 stimulates CD36 expression.7 The underlying mechanisms of the conflicting results are unknown and our current study showed that siRNA against IL-10 did not abolish the downregulation of CD36 by IL-33 but further decreased CD36 expression in MFCs, which at least indicates that IL-10 does not play a role in IL-33-induced decreased CD36 levels.
The effects of IL-10 on MFC formation are highly controversial and the underlying mechanisms may be due to different cells and the stimulations given.5, 7 However, regardless of the exact effect of IL-10 on lipid accumulation in MFCs, studies have consistently shown that IL-10 prevents cholesterol over-loading in MFCs, which is a PPAR-γ-dependent pathway.5, 7 Interestingly, it is important to note that an in vivo study showed that IL-33 promoted IL-10 production and activated PPAR-γ.9 As such, the current findings that siRNA-IL10 abolished IL-33-induced cholesterol could also be explained by the attenuation PPAR-γ activation by IL-10 siRNA.
Several limitations of the current study should be acknowledged. First, a lack of atherosclerotic coronary plaques restricted the demonstration of IL-33 and IL-10 alterations in local atherosclerotic lesions; therefore, we used circulating IL-33 and IL-10 instead. However, these cytokines may not necessarily represent the micro-environment within atherosclerotic plaques. Second, we did not explore all of the transcription factors and potential target sequences involved in the STAT3–DNA interactions involved in IL-33-induced IL-10 expression. In the present study, we chose the target transcription factors mainly according to the luciferase activity results, published items (involved in EKR 1/2, transcription factors and IL-10 expression) and the predictions from two databases. Therefore, we focused on the predicted sequences and STAT3; however, this approach does not exclude the possibility of other transcription factors and sequences having a role in this process. For example, within the −2000 to −1752 bp, the JASPAR results, but not Genomatix Matlnspector results, despite a low score, predicted two binding sites for Sp1, which was previously shown to bind −96 to −76 bp and regulate IL-10 expression. For STAT3–DNA interactions, we showed that the distal region may play the most important role, but the classical proximal region may also have an effect; however, we did not examine this aspect in details. Therefore, the present study does not exclude a role of other transcription factors and STAT3–DNA interaction sequences in the regulation of IL-10 by IL-33 in MFCs.
Despite these limitations, to the best our knowledge, this study is the first to find that IL-33 induces IL-10 production by macrophages, which helps IL-33 to increase ABCA1 expression and benefit MFCs. Furthermore, we demonstrated that the IL-33-induced IL-10 production is transcription-dependent and identified, at least in part, transcription factor and target sequences that are involved. More studies are needed to explore the effects between IL-33, IL-10 and foam cells in vivo and a more broad mechanism involving IL-33 and IL-10 may be helpful to explain the effects of IL-33 on MFCs in more detail.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005; 23: 479–490.
Kurowska-Stolarska M, Hueber A, Stolarski B, McInnes IB . Interleukin-33: a novel mediator with a role in distinct disease pathologies. J Intern Med 2011; 269: 29–35.
Miller AM, Xu D, Asquith DL, Denby L, Li Y, Sattar N et al. IL-33 reduces the development of atherosclerosis. J Exp Med 2008; 205: 339–346.
McLaren JE, Michael DR, Salter RC, Ashlin TG, Calder CJ, Miller AM et al. IL-33 reduces macrophage foam cell formation. J Immunol 2010; 185: 1222–1229.
Rubic T, Lorenz RL . Downregulated CD36 and oxLDL uptake and stimulated ABCA1/G1 and cholesterol efflux as anti-atherosclerotic mechanisms of interleukin-10. Cardiovasc Res 2006; 69: 527–535.
Yang H, Chen S, Tang Y, Dai Y . Interleukin-10 down-regulates oxLDL induced expression of scavenger receptor A and Bak-1 in macrophages derived from THP-1 cells. Arch Biochem Biophys 2011; 512: 30–37.
Han X, Kitamoto S, Lian Q, Boisvert WA . Interleukin-10 facilitates both cholesterol uptake and efflux in macrophages. J Biol Chem 2009; 284: 32950–32958.
Mei CL, Chen ZJ, Liao YH, Wang YF, Peng HY, Chen Y . Interleukin-10 inhibits the down-regulation of ATP binding cassette transporter A1 by tumour necrosis factor-alpha in THP-1 macrophage-derived foam cells. Cell Biol Int 2007; 31: 1456–1461.
Miller AM, Asquith DL, Hueber AJ, Anderson LA, Holmes WM, McKenzie AN et al. Interleukin-33 induces protective effects in adipose tissue inflammation during obesity in mice. Circ Res 2010; 107: 650–658.
Brunner SM, Schiechl G, Falk W, Schlitt HJ, Geissler EK, Fichtner-Feigl S . Interleukin-33 prolongs allograft survival during chronic cardiac rejection. Transpl Int 2011; 24: 1027–1039.
Saraiva M, O’Garra A . The regulation of IL-10 production by immune cells. Nat Rev Immunol 2010; 10: 170–181.
Liu G, Ng H, Akasaki Y, Yuan X, Ehtesham M, Yin D et al. Small interference RNA modulation of IL-10 in human monocyte-derived dendritic cells enhances the Th1 response. Eur J Immunol 2004; 34: 1680–1687.
Xie ZQ, Liang G, Zhang L, Wang Q, Qu Y, Gao Y et al. Molecular mechanisms underlying the cholesterol-lowering effect of Ginkgo biloba extract in hepatocytes: a comparative study with lovastatin. Acta Pharmacol Sin 2009; 30: 1262–1275.
Rosenblat M, Oren R, Aviram M . Lysophosphatidylcholine (LPC) attenuates macrophage-mediated oxidation of LDL. Biochem Biophys Res Commun 2006; 344: 1271–1277.
Wang Y, Wang X, Sun M, Zhang Z, Cao H, Chen X . NF-kB activity-dependent P-selectin involved in ox-LDL-induced foam cell formation in U937 cell. Biochem Biophys Res Commun 2011; 411: 543–548.
Maroof A, Beattie L, Zubairi S, Svensson M, Stager S, Kaye PM . Post-transcriptional regulation of II10 gene expression allows natural killer cells to express immunoregulatory function. Immunity 2008; 29: 295–305.
Yao Y, Xu Q, Kwon MJ, Matta R, Liu Y, Hong SC et al. ERK and p38 MAPK signaling pathways negatively regulate CIITA gene expression in dendritic cells and macrophages. J Immunol 2006; 177: 70–76.
Ma W, Lim W, Gee K, Aucoin S, Nandan D, Kozlowski M et al. The p38 mitogen-activated kinase pathway regulates the human interleukin-10 promoter via the activation of Sp1 transcription factor in lipopolysaccharide-stimulated human macrophages. J Biol Chem 2001; 276: 13664–13674.
Kube D, Platzer C, von KA, Straub H, Bohlen H, Hafner M et al. Isolation of the human interleukin 10 promoter. Characterization of the promoter activity in Burkitt’s lymphoma cell lines. Cytokine 1995; 7: 1–7.
Lucas M, Zhang X, Prasanna V, Mosser DM . ERK activation following macrophage FcgammaR ligation leads to chromatin modifications at the IL-10 locus. J Immunol 2005; 175: 469–477.
Foey AD, Feldmann M, Brennan FM . CD40 ligation induces macrophage IL-10 and TNF-alpha production: differential use of the PI3K and p42/44 MAPK-pathways. Cytokine 2001; 16: 131–142.
Sanada S, Hakuno D, Higgins LJ, Schreiter ER, McKenzie AN, Lee RT . IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J Clin Invest 2007; 117: 1538–1549.
Yang Z, Mosser DM, Zhang X . Activation of the MAPK, ERK, following Leishmania amazonensis infection of macrophages. J Immunol 2007; 178: 1077–1085.
Liu BS, Cao Y, Huizinga TW, Hafler DA, Toes RE . TLR-mediated STAT3 and ERK activation controls IL-10 secretion by human B cells. Eur J Immunol 2014; 44: 2121–2129.
Saraiva M, Christensen JR, Veldhoen M, Murphy TL, Murphy KM, O’Garra A . Interleukin-10 production by Th1 cells requires interleukin-12-induced STAT4 transcription factor and ERK MAP kinase activation by high antigen dose. Immunity 2009; 31: 209–219.
Hong GH, Kwon HS, Lee KY, Ha EH, Moon KA, Kim SW et al. hMSCs suppress neutrophil-dominant airway inflammation in a murine model of asthma. Exp Mol Med 2017; 49: e288.
Chanteux H, Guisset AC, Pilette C, Sibille Y . LPS induces IL-10 production by human alveolar macrophages via MAPKinases- and Sp1-dependent mechanisms. Respir Res 2007; 8: 71.
Liu Y, Chen Q, Song Y, Lai L, Wang J, Yu H et al. MicroRNA-98 negatively regulates IL-10 production and endotoxin tolerance in macrophages after LPS stimulation. FEBS Lett 2011; 585: 1963–1968.
Sharma A, Kumar M, Aich J, Hariharan M, Brahmachari SK, Agrawal A et al. Posttranscriptional regulation of interleukin-10 expression by hsa-miR-106a. Proc Natl Acad Sci USA 2009; 106: 5761–5766.
Zhao N, Li S, Wang R, Xiao M, Meng Y, Zeng C et al. Expression of microRNA-195 is transactivated by Sp1 but inhibited by histone deacetylase 3 in hepatocellular carcinoma cells. Biochim Biophys Acta 2016; 1859: 933–942.
Yan L, Yu J, Tan F, Ye GT, Shen ZY, Liu H et al. SP1-mediated microRNA-520d-5p suppresses tumor growth and metastasis in colorectal cancer by targeting CTHRC1. Am J Cancer Res 2015; 5: 1447–1459.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation for Young Scientists of China to ZH-F (Grant No. 81300071) and MQ-Q (Grant No. 81301038), the Natural Science Foundation of Guangdong Province of China to LY-Q (Grant No. 2014A030313049) and LN-S (Grant No. 2014A030313655); the Science and Technology Program of Guangzhou City of China to ZH-F (Grant No. 201510010048) and LN-S (Grant No. 201510010050), and the Fundamental Research Funds for the Central Universities to ZH-F (Grant No. 13ykpy30).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Experimental & Molecular Medicine website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Zhang, HF., Wu, MX., Lin, YQ. et al. IL-33 promotes IL-10 production in macrophages: a role for IL-33 in macrophage foam cell formation. Exp Mol Med 49, e388 (2017). https://doi.org/10.1038/emm.2017.183
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2017.183
This article is cited by
-
Curcumin attenuates inflammation of Macrophage-derived foam cells treated with Poly-L-lactic acid degradation via PPARγ signaling pathway
Journal of Materials Science: Materials in Medicine (2022)
-
IL-33 and its decoy sST2 in patients with Alzheimer’s disease and mild cognitive impairment
Journal of Neuroinflammation (2020)
-
Inflammatory markers and bone health in postmenopausal women: a cross-sectional overview
Immunity & Ageing (2019)
-
The interleukin-33-mediated inhibition of expression of two key genes implicated in atherosclerosis in human macrophages requires MAP kinase, phosphoinositide 3-kinase and nuclear factor-κB signaling pathways
Scientific Reports (2019)
