
Abstract
Pulmonary arterial hypertension (PAH) is a rare but progressive and currently incurable disease, which is characterized by vascular remodeling in association with muscularization of the arterioles, medial thickening and plexiform lesion formation. Despite our advanced understanding of the pathogenesis of PAH and the recent therapeutic advances, PAH still remains a fatal disease. In addition, the susceptibility to PAH has not yet been adequately explained. Much evidence points to the involvement of epigenetic changes in the pathogenesis of a number of human diseases including cancer, peripheral hypertension and asthma. The knowledge gained from the epigenetic study of various human diseases can also be applied to PAH. Thus, the pursuit of novel therapeutic targets via understanding the epigenetic alterations involved in the pathogenesis of PAH, such as DNA methylation, histone modification and microRNA, might be an attractive therapeutic avenue for the development of a novel and more effective treatment. This review provides a general overview of the current advances in epigenetics associated with PAH, and discusses the potential for improved treatment through understanding the role of epigenetics in the development of PAH.
Similar content being viewed by others


Introduction
Pulmonary hypertension (PH) is a disorder in the lung vasculatures including the pulmonary artery, pulmonary vein or pulmonary capillaries, resulting in an increase of blood pressure followed by heart failure.1 After the clinical classification of PH into primary and secondary PH at the first meeting held by the World Health Organization (WHO) in 1973, the categories of PH were continuously subdivided more precisely, until reestablishment according to the presence of the identified causes at the 5th World Symposium of Pulmonary Hypertension held in Nice, France, in 2013. The recent updated classification of PH presents five WHO groups as follows: (i) WHO group 1, pulmonary arterial hypertension (PAH); (ii) WHO group 2, pulmonary hypertension due to left heart disease; (iii) WHO group 3, pulmonary hypertension due to lung diseases and/or hypoxia; (iv) WHO group 4, chronic thromboembolic pulmonary hypertension; and (v) WHO group 5, pulmonary hypertension with unclear multifactorial mechanisms. Each group was also further subdivided by its genetic or pathological causes.2 PAH, the WHO group 1, is a disorder of the pulmonary arterioles, resulting in increased blood pressure followed by right ventricular heart failure, and characterized by the absence of the common causes of PH, which include chronic liver and thromboembolic diseases. The pathogenic events of PAH arise from the hyperproliferation of pulmonary vascular cells, such as pulmonary artery endothelial cells (PAECs) and pulmonary artery smooth muscle cells (PASMCs), which in turn causes neointima formation in the small pulmonary arteries.3 Although rare, occurring at only 2.4–7.6 cases per million per year, PAH is a progressive disease leading to an incident mortality rate of ~15% within 1 year of diagnosis. Moreover, the mortality rate of PAH was reported in 2012, to be 51% within 7 years of diagnosis.4, 5
PAH is a complex disease with multiple etiologies and may be mediated by the interplay of genetic background, epigenetic changes and pathobiological environmental factors, which explains the great variability in susceptibility6 (Figure 1). Therefore, the defining molecular mechanisms involved in the pathogenesis of PAH may arise from various aspects due to the multiple etiologies and disease heterogeneity. Emerging evidence has demonstrated the importance of epigenetics in the pathogenesis of PAH.6, 7, 8, 9 Epigenetics is defined as all heritable changes in gene expression that are not related to changes in the underlying DNA sequence.10 To date, the cell-signaling abnormalities, and environmental and genetic mechanisms involved in PAH pathogenesis, have been well studied. However, despite advances in epigenetics technology such as genome-scale DNA methylation analysis, few studies have yet been performed on the epigenetics associated with PAH pathogenesis. The three main types of epigenetic regulation are DNA methylation, histone modification and microRNA (miRNA).11 Although many miRNAs associated with PAH have been elucidated, the involvement of epigenetic regulation via methylation and histone modification in the pathogenesis of PAH remains in critical need of investigation. Our efforts for understanding the initiation and progression of PAH via epigenetics research may provide new insights to identify novel targets for treatment. This review will introduce the current understanding of the epigenetics associated with PAH pathobiology and discuss the possible epigenetic modulations involved in progression of PAH.

Proposed multifactorial pathogenesis of pulmonary arterial hypertension (PAH). This figure presents the complex nature of heritable PAH (HPAH) and idiopathic PAH (IPAH). In the case of HPAH, the major driver ‘primary hit’ maybe genetic mutation of HPA-associated genes. In many PAH patients, unknown or undetectable ‘secondary hit’ mechanisms such as epigenetic alteration, gender and other cardiovascular anomalies, as well as environmental factors, might cooperate in the progression of HPAH. Commonly, IPAH is caused by the combination effect of multiple cues such as non-heritable genetic or epigenetic variations, as well as environmental statuses.
Major mechanisms of epigenetic regulation
DNA methylation
Although most epigenetic changes are highly dynamic, depending on the cellular status, DNA methylation is comparably stable and can even be inherited by daughter cells. In the genome, methyl groups can be added by covalent interaction to adenosine or cytosine DNA nucleotides through the enzyme DNA methyltransferase (DNMT).12, 13 DNA methylation and demethylation processes govern diverse biological conditions, involving genomic imprinting, cellular differentiation, organ morphogenesis, cell reprogramming, X-chromosome inactivation, RNA splicing, transposon silencing and DNA repair.14 The CpG island, a region with a specific base sequence commonly located in gene promoters, can undergo particularly high rates of methylation, which is considered as a sign of genetic repression. Mechanistically, DNA methylation achieves transcriptional repression through three modes of action as follows: (1) direct masking of the binding site of transcription factors, caused by nucleosome compaction; (2) recruitment of transcriptional repressors; and (3) cross-interaction with histone modification mechanisms.15, 16 Conversely, the promoter regions of most active genes appear to be unmethylated (or hypomethylated). For example, oncogenes are generally hypermethylated, whereas tumor suppressor genes are unmethylated in cells with normal cellular status; this epigenetic state is reversed in the progression of cancer.17 Consequently, the detection of global changes in DNA methylation can be considered as a hallmark of cancer pathogenesis.18, 19 During embryonic development, organogenesis and general pathogenesis, functional genes can also be controlled spatiotemporally by DNA methylation mechanisms. Moreover, changes of the DNA methylation state are known to have close relations to a variety of human diseases and disorders including cancer, diabetes, immune disorders, cardiovascular diseases and cerebral ischemia, as well as genetically imprinted disorders, suggesting the strong possibility for therapeutic applications of this epigenetic mechanism.20 In such diseases, detection of the epigenome state through analysis of the global DNA methylation state might be important, as epigenetic changes can be considered as biomarkers for interpretation of pathogenic status. The fact that the alteration of DNA methylation by environmental input can be observed is a very interesting biological phenomenon. In addition to genetic cues of pathogenic abnormality, this can lead to a unique synergetic condition through accumulation of hyper- or hypo-DNA methylation, causing it to be regarded as a ‘second hit’ in many disease conditions. Alcohol, tobacco, cocaine and many drugs are capable of modifying the global DNA methylation in target genomic regions of sensitive organs, especially in conditions of pregnancy.21, 22
Histone modification
Double-stranded DNA is highly packed with incorporation of histones and is organized as the nucleosome to build chromatin in eukaryotic cell nuclei. In nature, five histone families, H1/H5, H2A, H2B, H3 and H4, were found to be incorporated into chromatin. Although the H1/H5 histone family has the role of a linker, the other histones function to form a core structure with the DNA strand. In biological events, the posttranscriptional modification of these histones is related to diverse functional aspects including the regulation of gene transcription, DNA repair process, chromosome condensation and meiosis, as well as genetic imprinting.23, 24 Similar to other proteins, histones are diversely modified by posttranslational modification through the following mechanisms: methylation, acetylation, ubiquitination, SUMOylation, phosphorylation and ADP-ribosylation. These constitute the complex ‘Histone Code,’ which is determined by the combinational modifications of histone.25, 26, 27 In particular, histone methylation and acetylation are closely linked with transcriptional gene regulation, which is regarded as one of the major epigenetic events and is related with many pathological conditions in the progression of cardiovascular disease, including PAH.9, 26 Many of the lysine (K) residues of histone H3 such as H3K4, H3K9, H3K27, H3K36 and H3K79 are commonly methylated by adding one, two or three methyl groups, respectively. Although the methylation of histone H3K9 and H3K27 is thought to be a code for transcriptional repression, the methylation of histone H3K4, H3K36 and H3K79 is able to induce gene transcription.28 A variety of histone methyltransferases and histone demethylases are involved in controlling the state of histone methylation. In addition to histone methylation, the acetylation of histone H3, especially of H3K9, H3K14 and H3K27, can also function as an active transcriptional code. These biological processes are governed by histone acetyltransferase and histone deacetylase (HDAC).29, 30 Specifically, HDAC removes the acetyl groups from lysine residues on the histone, causing chromatin to become tightly packaged, and represses gene expression. In humans, 18 HDACs within 4 classes have been discovered, including class I, IIa, IIb, III and IV. Except for class III HDACs, which are known as sirtuins (SIRT1-7), the other three classes of HDACs are zinc-dependent enzymes, which are commonly targets of small chemical HDAC inhibitors.31, 32 Recent evidence has suggested that inflammation widely contributes to cardiovascular pathogenesis, including the pathogenesis of PAH.33 Importantly, treatment with HDAC inhibitors at a low dose can attenuate the inflammatory response in chronic cardiovascular conditions, providing the potential functional mechanism of HDAC inhibitors in therapeutic treatment.34
MicroRNAs
miRNAs are endogenous, short-length (20–24 nucleotides), noncoding RNAs that are involved in the posttranscriptional fine-tuning of gene expression, typically through binding to the 3′-untranslated region, to affect the stability and translation of target mRNAs. miRNAs are expressed in multicellular organisms and are highly conserved between species.35 As one miRNA has the capacity to target multiple mRNAs, elaborate regulation of miRNA expression is fundamental to maintaining homeostasis in living organisms. Accordingly, altered expression of miRNAs can cause pathogenic conditions such as PAH.35 In addition, emerging studies have shown that epigenetic modifications by DNA methylation and histone modification can also regulate the expression of miRNAs in a transcriptional manner, suggesting the therapeutic potential of HDAC and DNMT modulators for regulating miRNA expression for the treatment of PAH.9, 36
Epigenetic alterations in PAH
DNA methylation and histone modification in PAH
Given the exacerbated severity of PAH by the interplay of complex genetic and/or epigenetic changes, identification of novel therapeutic avenues via investigation of the epigenetic mechanisms involved in the pathogenesis of PAH might be of growing interest. However, few studies have identified the role of epigenetic modifications such as altered DNA methylation and histone modification in association with PAH pathogenesis.7, 8, 9
Superoxide dismutase-2 (SOD2) is a member of the iron/manganese SOD family, which catalyzes the dismutation of superoxide into hydrogen peroxide and diatomic oxygen, and has a critical role in vascular functions.37 Several lines of study have implicated SOD2 in the development of PAH.8, 38 One study showed that adenovirus-mediated gene transfer of SOD to the lung ameliorated monocrotaline-induced PH in rats, suggesting the involvement of increased oxidative stress in the pathogenesis of PAH and the therapeutic potential of antioxidants for treatment.38 Epigenetic changes by histone modification are a key mechanism for regulation of cell proliferation and survival. The first demonstration of aberrant epigenetic changes in PAH pathogenesis showed no mutation in the SOD2 gene, whereas tissue-specific, methylation-induced SOD2 deficiency increased the proliferation and decreased the apoptosis of PASMC, while also impairing redox signaling. Conversely, SOD augmentation restores experimental PAH, suggesting therapeutic benefits of epigenetic modification.8
Zhao et al.7 also demonstrated that epigenetic modifications, through histone acetylation, are implicated in the development of PAH. The levels of HDAC1 and HDAC5 were higher in lungs from both PAH patients and a PH rat model than in control groups, and HDAC inhibitors were found to exert antiproliferative and anti-inflammatory effects on vascular cells. The authors also demonstrated that HDAC inhibitors, including suberoylanilide hydroxamic acid and vorinostat, ameliorate the phenotype of PH rat models, indicating that increased HDAC activity leads to the pathological condition of PH.7 Another investigation by Wang et al.36 showed that miR-124 has an important role in maintaining homeostasis in fibroblasts and is involved in the pathogenesis of PAH. miR-124 significantly inhibits proliferation, migration and expression of monocyte chemotactic protein-1 in pulmonary vascular fibroblasts. In addition, the expression of miR-124 is significantly decreased in the fibroblasts of patients with PAH. Interestingly, the decrease in miR-124 was restored by treatment with HDAC inhibitors, but not by 5-aza-deoxycytidine in hypertensive fibroblasts.36 These studies suggest therapeutic potential for HDACs inhibitors in the treatment of PAH.
Myocyte enhancer factor 2 (MEF2) is a family of transcription factors, which are known to have an important role in control of the expression of genes involved in cellular differentiation and embryonic development. There are four members of the MEF2 family: MEF2A, MEF2B, MEF2C and MEF2D. Among them, MEF2A and MEF2C are highly expressed in endothelial cells. Endothelial-cell-specific MEF2C-deficient mice showed reduced retinal vessel loss and decreased endothelial apoptosis, suggesting that MEF2 is a key endothelial homeostatic transcription factor in the vasculature.39, 40, 41, 42 In light of the function of MEF2 in endothelial cells, the most recent study by Kim et al.9 found a novel role for the transcription factor MEF2 in maintaining homeostasis in the pulmonary vasculature. MEF2 activity was also found to be impaired in PAH PAECs through excess nuclear accumulation of HDAC4 and HDAC5. The impaired MEF2 activity leads to the downregulation of target genes involved in pulmonary vascular homeostasis, including miR-424 and 503, connexins 37 and 40, and Krűppel-like factors 2 and 4 (Kim et al.9). Especially, miR-424 and 503 have been revealed as key miRNAs in maintaining homeostasis, the disruption of which lead to the pathogenesis of PAH.43 The authors also demonstrated that selective, pharmacological inhibition of class IIa HDACs using MC1568 restored the impaired MEF2 activity in PAH PAECs and rescued the experimental monocrotaline and SU-5416/hypoxia (SUGEN) PH models, while restoring targets such as miR-424 and 503 (Kim et al.9). The reports described above provide direct evidence of the role of epigenetics in PAH pathogenesis, contributing to the vascular pathology of PAH.
miRNAs in PAH
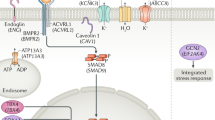
As PAH is a complex disease with multiple etiologies, miRNAs may be key candidates for more effective treatment, due to their capacity to coordinately regulate various signaling pathways associated with PAH such as bone morphogenetic protein (BMP) signaling, apelin (APLN) and apelin receptor (APLNR) signaling and hypoxia-related signaling, via the targeting of multiple mRNAs.1, 44, 45, 46 Thus, the abnormal expression and dysregulation of miRNAs contributes to the pathogenesis of PAH. A significant amount of study has been devoted to elucidating the roles of miRNAs, as one epigenetic mechanism involved in the pathogenesis of PAH, through which many altered miRNAs have been identified.1, 35 The PAH-related miRNAs and signaling mechanisms, including their target mRNAs, are summarized in Table 1 and Figure 2.

MicroRNA (miRNA) regulatory pathways implicated in the pathogenesis of pulmonary arterial hypertension (PAH). This figure provides an overview of the signaling pathways regulated by miRNAs, which are involved in the pathogenesis of PAH. Aberrant expression of miRNAs in pulmonary vascular cells such as pulmonary artery endothelial cells (PAECs), pulmonary artery smooth muscle cells (PASMCs) and fibroblasts by pathological factors leads to altered signaling pathways and contributes to the pathogenesis of PAH.
Possible epigenetic alteration of genes associated with PAH
Components of BMP signaling
BMP signaling is a part of the transforming growth factor (TGF)-β superfamily, which consists of TGF-β, BMP, activin and growth and differentiation factor (GDF) signaling.47, 48 The recent circumstantial evidence suggests that BMP signals are involved in the development of endothelium and maintenance of blood vessel homeostasis.49, 50, 51 In particular, several human genetic mutations have been closely linked with pathogenic conditions related to the cardiovascular system such as hereditary hemorrhagic telangiectasia and PH.52, 53, 54 The following section will discuss the potential possibility of epigenetic regulation of the major components involved in the BMP signaling cascade and the subsequent influence on PAH progression.
BMP type I receptors
BMP type I receptor (BMPR1) directly binds to BMP ligands such as BMP2, 4, 6, 9 and 10, and is activated by phosphorylation through subsequent formation of tetraheteromeric complexes with BMP receptor type II (BMPR2). In genetic screening experiments in PH (or PAH) patients, mutations in BMPR1 genes, including activin A receptor type II-like 1 (ACVRL1/ALK1) and BMP receptor type-1B (BMPR1B/ALK6), were identified to be involved in predisposal to PAH.55, 56 Moreover, the downregulation of BMP receptor type-1A (BMPR1A/ALK3), mediated by angiopoietin-1 signaling in the lung tissue, was suggested to be a cause of nonfamilial PH.57 Although direct evidence of epigenetic regulation of the BMPR1 genes has not yet been finely addressed in cardiovascular pathogenesis, there may be considerable functional contributions of epigenetic mechanisms to these genes. For instance, the expression of ACVRL1/ALK1, which is dominantly expressed in endothelial cells, is controlled by CpG island methylation, mediated by transcription factor Sp1 on the ACVRL1/ALK1 promoter region, suggesting possible involvement of the epigenetic mechanism of ACVRL1/ALK1 in PAH.58 In recent times, miR-656 was reported to directly target BMPR1A/ALK3 in glioma cell lines.59 In the case of BMPR1B/ALK6, it was suggested that the expression of this gene can be tightly regulated through the DNA methylation status of the species-conserved 5′-CpG island in the promoter region. The hypermethylation of this region silences BMPR1B expression in human glioblastoma tumor-initiating cells and results in inhibition of normal cell differentiation and subsequent tumorigenicity. Moreover, treatment with a demethylation agent such as 5-aza-2′-deoxycytidine or genetic inhibition of enhancer of zeste homolog 2, which functions in the methylation of CpG islands by recruiting DNMT, restored the expression of BMPR1B in human glioblastoma tumor-initiating cells.60 Collectively, these epigenetic regulations of BMPR1 genes may also be involved in PAH pathogenesis, and require further elucidation, as these biological phenomena have not yet been described in the cardiovascular system.
BMP receptor type II
The unique type II serine/threonine kinase receptors in components of the TGF-β superfamily, involving TGF-β, BMP and activin signaling, determine the downstream signaling cascade. BMPR2 has a unique signaling capacity only for BMP signal transduction, chiefly relayed by SMAD1, 5 and 8 (Ehrlich et al.47). Around 70% of familial PAH patients were found to have heritable genetic mutations of BMPR2. Moreover, genetic alteration of BMPR2 was found in about 10% to 40% of idiopathic PAH (IPAH) patients.61, 62 However, the potential susceptibility to genetic alteration of BMPR2 in heritable PAH and IPAH is relatively low, as ~20% of individuals have heritable mutations, suggesting incomplete penetrance of PAH and the requirement for another secondary cue to promote development of PAH.63 The germline mutation of BMPR2 is regarded as the most common causative factor and can be targeted for primary therapeutic application in advance. Currently, the involvement of epigenetic alteration or regulation of BMPR2 in PAH progression has not been clearly defined; however, there are several examples of the involvement of epigenetic regulation of BMPR2 in various pathological conditions including cardiovascular disorders. For example, miR-17-5p and miR-20a were reported to be closely related to BMPR2 expression in PH pathogenesis, having a role in BMPR2 downregulation in PAEC and PASMC64, 65 (Figure 2). On the other hand, the involvement of other epigenetic regulations such as histone modification and DNA methylation of BMPR2 are largely unknown and little have been suggested. In recent times, the CpG island on the BMPR2 promoter was found to be hypermethylated in scleroderma endothelial cells, whereas treatment with a DNMT inhibitor and/or HDAC inhibitor reversed the enhanced apoptosis of the cells, illuminating the possible contribution of abnormal DNA methylation in scleroderma pathogenesis.66 These epigenetic contributions might also have a relation to other cardiovascular disorders, including PAH, and should be addressed further in PAEC and PASMC beds, to reveal possible roles in PAH.
Endoglin: co-receptor of TGF-β/BMP signaling
Although endoglin was originally identified as a TGF-β receptor, it is now regarded as a co-receptor for TGF-β and BMP signaling. In particular, the receptor activity of ALK1, which dominantly binds with the BMP9 or 10 ligands, can be modified by the presence or absence of endoglin. In human cardiovascular pathogenesis, endoglin has been identified as the causal gene for hereditary hemorrhagic telangiectasia type I, classified by the clinical presence of recurrent epistaxis and spontaneous arterial venous malformation, sometimes followed by PH or PAH.67 Several epigenetic mechanisms regulating the expression of endoglin were reported in tumor conditions. For example, endoglin was reported to be critically downregulated in numerous esophageal squamous cell carcinoma tissues, due to promoter hypermethylation of the endoglin locus. Treatment with a demethylation agent restored the endoglin expression in carcinoma cell lines, suggesting the potential epigenetic regulation of the endoglin promoter.68 Moreover, several miRNAs are known to target endoglin in endothelial cells or cardiac myocytes. In particular, miR-208a repressed endoglin expression in the heart, whereas miR-370 was negatively correlated with endoglin expression in endometrioid ovarian cancer cells.69, 70
SMADs: main mediators of BMP signaling
Several receptor-mediated SMADs such as SMAD1, 5, 8 or 9 and a common-mediator SMAD, SMAD4, are involved in canonical BMP signal transduction in various biological processes including the development and homeostasis of the cardiovascular system. Some functional and genetic involvements of these signaling mediators have been reported. The truncating mutation of SMAD9 was identified in familial PAH patients, and genetic variants in SMAD1 and SMAD4 were identified as a causative factor for IPAH.71, 72 Interestingly, the pathological results from a SMAD9 knockout mouse model strongly support the functional involvement of SMAD genes in the progression of human PAH.56 It was demonstrated that the translation of SMAD genes is epigenetically modified by multiple sets of miRNAs in various cell types such as endothelial and vascular smooth muscle cells, as well as mesenchymal stem cells, for bone differentiation. For example, miR-4448, -4708 and -4773 target SMAD1 and SMAD4 transcripts, to induce osteoblast differentiation from mesenchymal stem cells.73 Moreover, miR-26a, -30b and -205 are more closely involved in the cardiovascular system. miR-30b represses SMAD1 expression in aortic valve interstitial cells and its reduction might lead to calcific aortic valve disease.74 Several reports in the literature have suggested that SMAD1 and SMAD4 are common targets of miR-205 and -26a in endothelial and vascular smooth muscle cells, respectively, which are major contributors to lung homeostasis.75, 76 The translation of SMAD5 can be repressed by miR-155 and its similar sequence homolog, miR-K12-11, which is encoded by Kaposi’s sarcoma-associated herpesvirus.77, 78 Although the contribution of miRNAs targeting multiple SMAD genes in PAH has not yet been evaluated clearly, the possible involvement may be considerable.
Caveolin-1
Caveolin-1 is a membrane protein, which has the role of forming caveolae, and mainly functions in the endocytosis.79 In recent times, it was discovered that two frameshift mutations, a c.474delA and c.473delC, in the highly conserved C terminus amino acid sequence of caveolin-1 are related to familial PAH and IPAH.63 Interestingly, several reports suggest that the caveolae is able to control multiple signaling inputs in the endothelium, including BMPR2 and nitric oxide (NO) signaling.80, 81 Contributions of epigenetic DNA methylation to the expression of caveolin-1 have been suggested in adipocyte differentiation and soft tissue sarcoma. In particular, methylation of the promoter, exon1 and first intron region of caveolin-1 is extensively removed, inducing strong expression of caveolin-1 during adipocyte differentiation.82 Moreover, epigenetic regulation of caveolin-1 by multiple miRNAs such as a miR-133a, 802, 103, 107, 199a-3p, 199a-5p, 203 and 124 has been reported in diverse biological environments, including in insulin signaling, tumor cell proliferation and migration, and kidney homeostasis.83, 84, 85, 86, 87 Although the direct involvement of epigenetic regulation of caveolin-1 has not been revealed in the cardiovascular system, some of the suggested epigenetic mechanisms listed above may also be involved in cardiovascular pathogenesis, including PAH.
KCNK3 potassium channel
KCNK3 (Potassium channel subfamily K member 3) is a two-pore potassium channel majorly expressed in PASMC. In hypoxic conditions, KCNK3 is responsible for the control of membrane potential homeostasis. Currently, six cases of missense mutation (T8K, G97R, E182K, Y192C, G203D and V221L) have been reported in familial PAH and IPAH patients.88, 89 The mechanisms for epigenetic control of this gene in the cardiovascular system have not yet been elucidated, as this genetic alteration was only recently identified as a pathogenic cue of PAH.
Current therapies for PH
The current therapies for PAH patients commonly aim to reverse the imbalance of pulmonary vasoactive mediators. NO and prostacyclin are known vasodilators, whereas endothelin-1 (ET-1) and thromboxane A2 are known to function for vasoconstriction in PAEC and PASMC. Prostacyclin, a lipid produced by endothelial cells, causes vasodilatation and prevents the coagulation of platelets. It was reported in the 1990s that intravenous therapy with epoprostenol, a synthetic prostacyclin derivative, in combination with conventional therapies such as treatment with anticoagulants, cardiac glycosides and supplemental oxygen can reverse the severe progression of IPAH. Moreover, continuous infusion therapy with epoprostenol was found to be effective for several types of PAH patients, including those with PAH caused by systemic sclerosis or other connective tissue disease. To attenuate the progression of PAH, prostacyclin analogs are also available for inhalation as an aerosol formulation, or treatment as an orally active sustained release form.90, 91 Currently, many medical research groups are developing application methods for prostacyclin treatment, most of which are in clinical trials for multiple types of PAH patients including IPAH, connective tissue disease-associated PAH, HIV-associated PAH and congenital heart disease-associated PAH. Another target for PAH treatment is the ET receptor.91 ET is produced by endothelial cells and has a role in smooth muscle contraction. Mechanistically, ET binds to ET receptor type A (ET-A) and type B. Interestingly, the signaling output through ET receptor type B can cause a decline in the effects of ET-A, inducing mitogenic effects and vasoconstriction of vascular smooth muscle cells.92 It was suggested that in PAH progression, inhibition of ET-A signaling might relieve the pressure on the pulmonary artery by reducing the vasoconstriction of vascular smooth muscle cells.93 Several ET receptor antagonists such as bosentan, ambrisentan and macitentan have already been developed and approved, or are in trial for PAH treatment. Although the inhibition of ET-A activity by ET-A-selective or non-selective agents causes hepatotoxicity during PAH treatment, it provides sustained functional and physiological improvements to the pulmonary artery and relief of PAH symptoms.93, 94 It has long been regarded that NO is an important factor in vascular homeostasis. NO is released from the endothelium and modulates pulmonary and systemic vascular smooth muscle tones. Mechanistically, NO can activate soluble guanylate cyclase, resulting in increased levels of intracellular cyclic GMP. It was also reported that NO production is greatly reduced in PAH patients, whereas phosphodiesterase type 5, a major enzyme for cyclic GMP clearance, demonstrated increased expression, suggesting that the inhibition of phosphodiesterase type 5 might reverse PAH progression by relaxation of smooth muscle and inhibition of cell proliferation.95, 96 Several lines of phosphodiesterase type 5 inhibitors including sildenafil, tadalafil and vardenafil have been approved for PAH treatment. The improvement of PAH progression by phosphodiesterase type 5 inhibitor therapy has been well evaluated in multiple case reports, along with the tolerated levels of side effects such as headache, sudden visual loss and sudden hearing loss.97 Recently, several antiproliferative strategies using tyrosine kinase inhibitors were also developed. Platelet-derived growth factor receptor, epidermal growth factor receptor and fibroblast growth factor signal through tyrosine kinase receptors in the pathological remodeling of the vascular bed and have been targeted by multiple chemical inhibitors including imatinib, nilotinib, gefitinib, sorafenib and sunitinib, for reversal of PAH progression.98, 99 Although a greater understanding of the pathogenesis of PAH and PAH-specific therapies has led to significant advances, the disease is still incurable and ultimately fatal. Future therapies need to consider both regulation of pulmonary vascular tone and restoration of pulmonary vascular remodeling, to provide a more effective treatment for PAH through modulation of all the multifactorial factors such as epigenetic changes, pathobiological injurious events and genetic factors affecting PAH development.
Epigenetic modulation-based therapies for PH
Recent accumulating evidence has suggested that PAH is a complex and multifactorial disorder including overall endothelial malfunction, hyperproliferative vascular cells, anti-apoptosis of multiple compositions of the lung and proinflammatory response in the vascular bed, demonstrating the ‘maladaptive status’ of pulmonary arteries. This pathogenic concept gives credit to the environment and epigenetic alterations as a cause of PAH. Overall, it has been proposed that the progression and phenotypic variability of PAH may be affected by multifactorial factors such as genetic background, epigenetic changes, gender and pathobiological injurious events (virus, drug, toxin, hypoxia, inflammation and so on), whereas in a genetically susceptible background the effects of epigenetic changes and/or pathobiological injurious events may aggravate the disease severity6 (Figure 1). Interestingly, several experimental improvements through epigenetic modulations have been reported, which should be further evaluated for application to PAH patients. Among the epigenetic mechanisms, miRNAs have the capacity to regulate multiple target genes that may be involved in the pathogenesis of PAH, whereas dysregulation of miRNA expression could lead to the pathogenesis of PAH, which is a representative multifactorial disease. Alterations in the expression patterns of many miRNAs have been reported in PAH. Therefore, strategies to restore aberrant miRNA expression hold potential therapeutic value. Stable, efficient and nontoxic properties of miRNA mimics or inhibitors in vivo are an attractive therapeutic approach for the restoration of altered miRNA expression to physiological levels in PAH, while rescuing the expressions of target genes. However, a specific delivery strategy needs to be developed, to allow targeting of the lung vasculature to minimize off-target effects.1, 35 Pursuit of the transcriptional mechanisms of miRNA expression may also be a key field of research in PAH. Altered miRNA expression could be restored via modulation of the transcriptional mechanisms of miRNA expression. Several medical research groups are currently testing the therapeutic capability of modulation of HDAC in PAH pathogenesis, which are known to regulate cell proliferation and survival, as the results of both broad spectrum HDAC inhibitors and selective inhibitors demonstrated rescue of experimental PH in a rodent model.8, 9, 100 However, broad-spectrum HDAC inhibitors such as trichostatin A have displayed adverse clinical effects such as right ventricular dysfunction and marked induction of cellular apoptosis in coronary artery endothelial cells, indicating that particular attention is needed before their use as therapeutic agents in PAH subjects.101 Interestingly, most of the recent studies have indicated that the adverse effects observed could be avoided through selective inhibition of class IIa HDACs, suggesting that class IIa HDACs inhibitors may be a promising new treatment strategy for PAH, while allowing minimization of the side effects.9 The exploration of specific functional targets of histone modification and enhancement of selectivity of the chemical inhibitors to histone-modifying enzymes will be needed before application to PAH patients. Lastly, the use of DNMT inhibitors is another option to reverse the progressions of PAH associated with environmental and epigenetic alteration. For example, the down-expressional change of SOD2 was reported in a rat PAH model, which can be reversed by treatment with 5-azacytidine, a DNMT inhibitor. Although, the overall therapeutic concepts regarding epigenetic modulation in PAH are still being tested at the level of experimental conditions, gaining a precise biofunctional understanding of the genetic (for example, miRNAs) and chemical modulators (for example, inhibitors of histone and DNA-modifying enzymes) might open the door for novel treatment of PAH patients. However, careful attention should be given to epigenetic modulation-based therapies for PAH, to ensure their specificity.
Conclusion
Although epigenetic changes through DNA methylation and histone modification have been well defined in the pathogenesis of many human diseases such as cancer and peripheral hypertension,44, 102, 103 their application in the pathogenesis of PAH still remains in critical need of examination. A large amount of the research has been conducted on determining the role of miRNA and other epigenetic mechanisms.35 In order to gain a better understanding of the cause of PAH development, the intricate intersecting pathways between histone modification/DNA methylation and miRNA, which can be regulated by miRNA, histone modification or DNA methylation, need to be established. In addition, as epigenetic technology has been advanced to allow DNA methylation analysis, DNA/protein interaction analysis and chromatin accessibility/conformation assays, employment of such methods may allow the complex network of the many genes regulated by epigenetic mechanisms to be uncovered. The application of these epigenetic technologies to PAH research will provide a greatly improved understanding for the development of new drugs via novel targets and signaling pathways associated with PAH, and key insights into potential therapeutic strategies for PAH. Thus, future studies are needed to examine the role of epigenetics in the pathogenesis of PAH, as well as the therapeutic potential in experimental PH models.
References
Kim J . Apelin-APJ signaling: a potential therapeutic target for pulmonary arterial hypertension. Mol Cell 2014; 37: 196–201.
Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A et al. [Updated clinical classification of pulmonary hypertension]. Turk Kardiyol Dern Ars 2014; 42 (Suppl 1): 45–54.
Ma L, Chung WK . The genetic basis of pulmonary arterial hypertension. Hum Genet 2014; 133: 471–479.
Rosanio S, Pelliccia F, Gaudio C, Greco C, Keylani AM, D'Agostino DC . Pulmonary arterial hypertension in adults: novel drugs and catheter ablation techniques show promise? Systematic review on pharmacotherapy and interventional strategies. Biomed Res Int 2014; 2014: 743868.
Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD . An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 2012; 142: 448–456.
Tuder RM, Archer SL, Dorfmüller P, Erzurum SC, Guignabert C, Michelakis E et al. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol 2013; 62 (25 Suppl): D4–D12.
Zhao L, Chen CN, Hajji N, Oliver E, Cotroneo E, Wharton J et al. Histone deacetylation inhibition in pulmonary hypertension: therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation 2012; 126: 455–467.
Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation 2010; 121: 2661–2671.
Kim J, Hwangbo C, Hu X, Kang Y, Papangeli I, Mehrotra D et al. Restoration of impaired endothelial myocyte enhancer factor 2 function rescues pulmonary arterial hypertension. Circulation 2015; 131: 190–199.
Saco TV, Parthasarathy PT, Cho Y, Lockey RF, Kolliputi N . Role of epigenetics in pulmonary hypertension. Am J Physiol Cell Physiol 2014; 306: C1101–C1105.
Kim GH, Ryan JJ, Marsboom G, Archer SL . Epigenetic mechanisms of pulmonary hypertension. Pulm Circ 2011; 1: 347–356.
Movassagh M, Vujic A, Foo R . Genome-wide DNA methylation in human heart failure. Epigenomics 2011; 3: 103–109.
Liyanage VR, Jarmasz JS, Murugeshan N, Del Bigio MR, Rastegar M, Davie JR . DNA modifications: function and applications in normal and disease states. Biology (Basel) 2014; 3: 670–723.
Wu H, Zhang Y . Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell 2014; 156: 45–68.
Kass SU, Pruss D, Wolffe AP . How does DNA methylation repress transcription? Trends Genet 1997; 13: 444–449.
Piccolo FM, Fisher AG . Getting rid of DNA methylation. Trends Cell Biol 2014; 24: 136–143.
De Carvalho DD, Sharma S, You JS, Su SF, Taberlay PC, Kelly TK et al. DNA methylation screening identifies driver epigenetic events of cancer cell survival. Cancer Cell 2012; 21: 655–667.
Stirzaker C, Taberlay PC, Statham AL, Clark SJ . Mining cancer methylomes: prospects and challenges. Trends Genet 2014; 30: 75–84.
Beck S, Rakyan VK . The methylome: approaches for global DNA methylation profiling. Trends Genet 2008; 24: 231–237.
Kalish JM, Jiang C, Bartolomei MS . Epigenetics and imprinting in human disease. Int J Dev Biol 2014; 58: 291–298.
Ho SM, Johnson A, Tarapore P, Janakiram V, Zhang X, Leung YK . Environmental epigenetics and its implication on disease risk and health outcomes. ILAR J 2012; 53: 289–305.
Talikka M, Sierro N, Ivanov NV, Chaudhary N, Peck MJ, Hoeng J et al. Genomic impact of cigarette smoke, with application to three smoking-related diseases. Crit Rev Toxicol 2012; 42: 877–889.
Cheung P, Allis CD, Sassone-Corsi P . Signaling to chromatin through histone modifications. Cell 2000; 103: 263–271.
Zhou VW, Goren A, Bernstein BE . Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet 2011; 12: 7–18.
Kouzarides T . Chromatin modifications and their function. Cell 2007; 128: 693–705.
Bannister AJ, Kouzarides T . Regulation of chromatin by histone modifications. Cell Res 2011; 21: 381–395.
Bhaumik SR, Smith E, Shilatifard A . Covalent modifications of histones during development and disease pathogenesis. Nat Struct Mol Biol 2007; 14: 1008–1016.
Bannister AJ, Schneider R, Kouzarides T . Histone methylation: dynamic or static? Cell 2002; 109: 801–806.
Greer EL, Shi Y . Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 2012; 13: 343–357.
Chen T, Dent SY . Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat Rev Genet 2014; 15: 93–106.
Narlikar GJ, Sundaramoorthy R, Owen-Hughes T . Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell 2013; 154: 490–503.
Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M . Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov 2012; 11: 384–400.
El Chami H, Hassoun PM . Immune and inflammatory mechanisms in pulmonary arterial hypertension. Prog Cardiovasc Dis 2012; 55: 218–228.
Oudhoff MJ, Freeman SA, Couzens AL, Antignano F, Kuznetsova E, Min PH et al. Control of the hippo pathway by Set7-dependent methylation of Yap. Dev Cell 2013; 26: 188–194.
Lee A, McLean D, Choi J, Kang H, Chang W, Kim J . Therapeutic implications of microRNAs in pulmonary arterial hypertension. BMB Rep 2014; 47: 311–317.
Wang D, Zhang H, Li M, Frid MG, Flockton AR, McKeon BA et al. MicroRNA-124 controls the proliferative, migratory, and inflammatory phenotype of pulmonary vascular fibroblasts. Circ Res 2014; 114: 67–78.
Faraci FM, Didion SP . Vascular protection: superoxide dismutase isoforms in the vessel wall. Arterioscler Thromb Vasc Biol 2004; 24: 1367–1373.
Kamezaki F, Tasaki H, Yamashita K, Tsutsui M, Koide S, Nakata S et al. Gene transfer of extracellular superoxide dismutase ameliorates pulmonary hypertension in rats. Am J Respir Crit Care Med 2008; 177: 219–226.
De Val S, Anderson JP, Heidt AB, Khiem D, Xu SM, Black BL . Mef2c is activated directly by Ets transcription factors through an evolutionarily conserved endothelial cell-specific enhancer. Dev Biol 2004; 275: 424–434.
Subramanian SV, Nadal-Ginard B . Early expression of the different isoforms of the myocyte enhancer factor-2 (MEF2) protein in myogenic as well as non-myogenic cell lineages during mouse embryogenesis. Mech Dev 1996; 57: 103–112.
Xu Z, Gong J, Maiti D, Vong L, Wu L, Schwarz JJ et al. MEF2C ablation in endothelial cells reduces retinal vessel loss and suppresses pathologic retinal neovascularization in oxygen-induced retinopathy. Am J Pathol 2012; 180: 2548–2560.
Kang Y, Kim J, Anderson JP, Wu J, Gleim SR, Kundu RK et al. Apelin-APJ signaling is a critical regulator of endothelial MEF2 activation in cardiovascular development. Circ Res 2013; 113: 22–31.
Kim J, Kang Y, Kojima Y, Lighthouse JK, Hu X, Aldred MA et al. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat Med 2013; 19: 74–82.
Goto Y, Shinjo K, Kondo Y, Shen L, Toyota M, Suzuki H et al. Epigenetic profiles distinguish malignant pleural mesothelioma from lung adenocarcinoma. Cancer Res 2009; 69: 9073–9082.
McLean DL, Kim J, Kang Y, Shi H, Atkins GB, Jain MK et al. Apelin/APJ signaling is a critical regulator of statin effects in vascular endothelial cells—brief report. Arterioscler Thromb Vasc Biol 2012; 32: 2640–2643.
Chandra SM, Razavi H, Kim J, Agrawal R, Kundu RK, de Jesus Perez V et al. Disruption of the apelin-APJ system worsens hypoxia-induced pulmonary hypertension. Arterioscler Thromb Vasc Biol 2011; 31: 814–820.
Ehrlich M, Horbelt D, Marom B, Knaus P, Henis YI . Homomeric and heteromeric complexes among TGF-β and BMP receptors and their roles in signaling. Cell Signal 2011; 23: 1424–1432.
Kawabata M, Imamura T, Miyazono K . Signal transduction by bone morphogenetic proteins. Cytokine Growth Factor Rev 1998; 9: 49–61.
Kim JD, Kang H, Larrivée B, Lee MY, Mettlen M, Schmid SL et al. Context-dependent proangiogenic function of bone morphogenetic protein signaling is mediated by disabled homolog 2. Dev Cell 2012; 23: 441–448.
Wiley DM, Kim JD, Hao J, Hong CC, Bautch VL, Jin SW . Distinct signalling pathways regulate sprouting angiogenesis from the dorsal aorta and the axial vein. Nat Cell Biol 2011; 13: 686–692.
Kim JD, Lee HW, Jin SW . Diversity is in my veins: role of bone morphogenetic protein signaling during venous morphogenesis in zebrafish illustrates the heterogeneity within endothelial cells. Arterioscler Thromb Vasc Biol 2014; 34: 1838–1845.
David L, Feige JJ, Bailly S . Emerging role of bone morphogenetic proteins in angiogenesis. Cytokine Growth Factor Rev 2009; 20: 203–212.
Morse JH, Jones AC, Barst RJ, Hodge SE, Wilhelmsen KC, Nygaard TG . Mapping of familial primary pulmonary hypertension locus (PPH1) to chromosome 2q31-q32. Circulation 1997; 95: 2603–2606.
Nichols WC, Koller DL, Slovis B, Foroud T, Terry VH, Arnold ND et al. Localization of the gene for familial primary pulmonary hypertension to chromosome 2q31-32. Nat Genet 1997; 15: 277–280.
Morse JH, Deng Z, Knowles JA . Genetic aspects of pulmonary arterial hypertension. Ann Med 2001; 33: 596–603.
Chida A, Shintani M, Nakayama T, Furutani Y, Hayama E, Inai K et al. Missense mutations of the BMPR1B (ALK6) gene in childhood idiopathic pulmonary arterial hypertension. Circ J 2012; 76: 1501–1508.
Du L, Sullivan CC, Chu D, Cho AJ, Kido M, Wolf PL et al. Signaling molecules in nonfamilial pulmonary hypertension. N Engl J Med 2003; 348: 500–509.
Garrido-Martin EM, Blanco FJ, Fernandez-L A, Langa C, Vary CP, Lee UE et al. Characterization of the human activin-A receptor type II-like kinase 1 (ACVRL1) promoter and its regulation by Sp1. BMC Mol Biol 2010; 11: 51.
Guo M, Jiang Z, Zhang X, Lu D, Ha AD, Sun J et al. miR-656 inhibits glioma tumorigenesis through repression of BMPR1A. Carcinogenesis 2014; 35: 1698–1706.
Lee J, Son MJ, Woolard K, Donin NM, Li A, Cheng CH et al. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell 2008; 13: 69–80.
Liu D, Morrell NW . Genetics and the molecular pathogenesis of pulmonary arterial hypertension. Curr Hypertens Rep 2013; 15: 632–637.
Loyd JE . Pulmonary arterial hypertension: insights from genetic studies. Proc Am Thorac Soc 2011; 8: 154–157.
Austin ED, Loyd JE . The genetics of pulmonary arterial hypertension. Circ Res 2014; 115: 189–202.
Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M et al. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res 2009; 104: 1184–1191.
Brock M, Samillan VJ, Trenkmann M, Schwarzwald C, Ulrich S, Gay RE et al. AntagomiR directed against miR-20a restores functional BMPR2 signalling and prevents vascular remodelling in hypoxia-induced pulmonary hypertension. Eur Heart J 2014; 35: 3203–3211.
Wang Y, Kahaleh B . Epigenetic repression of bone morphogenetic protein receptor II expression in scleroderma. J Cell Mol Med 2013; 17: 1291–1299.
ten Dijke P, Goumans MJ, Pardali E . Endoglin in angiogenesis and vascular diseases. Angiogenesis 2008; 11: 79–89.
Jin Z, Zhao Z, Cheng Y, Dong M, Zhang X, Wang L et al. Endoglin promoter hypermethylation identifies a field defect in human primary esophageal cancer. Cancer 2013; 119: 3604–3609.
Wang BW, Wu GJ, Cheng WP, Shyu KG . MicroRNA-208a increases myocardial fibrosis via endoglin in volume overloading heart. PLoS ONE 2014; 9: e84188.
Chen XP, Chen YG, Lan JY, Shen ZJ . MicroRNA-370 suppresses proliferation and promotes endometrioid ovarian cancer chemosensitivity to cDDP by negatively regulating ENG. Cancer Lett 2014; 353: 201–210.
Nasim MT, Ogo T, Ahmed M, Randall R, Chowdhury HM, Snape KM et al. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum Mutat 2011; 32: 1385–1389.
Shintani M, Yagi H, Nakayama T, Saji T, Matsuoka R . A new nonsense mutation of SMAD8 associated with pulmonary arterial hypertension. J Med Genet 2009; 46: 331–337.
Kato RB, Roy B, De Oliveira FS, Ferraz EP, De Oliveira PT, Kemper AG et al. Nanotopography directs mesenchymal stem cells to osteoblast lineage through regulation of microRNA-SMAD-BMP-2 circuit. J Cell Physiol 2014; 229: 1690–1696.
Nigam V, Sievers HH, Jensen BC, Sier HA, Simpson PC, Srivastava D et al. Altered microRNAs in bicuspid aortic valve: a comparison between stenotic and insufficient valves. J Heart Valve Dis 2010; 19: 459–465.
Tabruyn SP, Hansen S, Ojeda-Fernández ML, Bovy N, Zarrabeitia R, Recio-Poveda L et al. MiR-205 is downregulated in hereditary hemorrhagic telangiectasia and impairs TGF-beta signaling pathways in endothelial cells. Angiogenesis 2013; 16: 877–887.
Liang H, Xu C, Pan Z, Zhang Y, Xu Z, Chen Y et al. The antifibrotic effects and mechanisms of microRNA-26a action in idiopathic pulmonary fibrosis. Mol Ther 2014; 22: 1122–1133.
Rai D, Kim SW, McKeller MR, Dahia PL, Aguiar RC . Targeting of SMAD5 links microRNA-155 to the TGF-beta pathway and lymphomagenesis. Proc Natl Acad Sci USA 2010; 107: 3111–3116.
Liu Y, Sun R, Lin X, Liang D, Deng Q, Lan K . Kaposi’s sarcoma-associated herpesvirus-encoded microRNA miR-K12-11 attenuates transforming growth factor beta signaling through suppression of SMAD5. J Virol 2012; 86: 1372–1381.
Kim J, Ahn S, Ko YG, Boo YC, Chi SG, Ni CW et al. X-linked inhibitor of apoptosis protein controls alpha5-integrin-mediated cell adhesion and migration. Am J Physiol Heart Circ Physiol 2010; 299: H300–H309.
Mineo C, Shaul PW . Regulation of eNOS in caveolae. Adv Exp Med Biol 2012; 729: 51–62.
Hartung A, Bitton-Worms K, Rechtman MM, Wenzel V, Boergermann JH, Hassel S et al. Different routes of bone morphogenic protein (BMP) receptor endocytosis influence BMP signaling. Mol Cell Biol 2006; 26: 7791–7805.
Palacios-Ortega S, Varela-Guruceaga M, Milagro FI, Martínez JA, de Miguel C . Expression of Caveolin 1 is enhanced by DNA demethylation during adipocyte differentiation. status of insulin signaling. PLoS ONE 2014; 9: e95100.
Nohata N, Hanazawa T, Kikkawa N, Mutallip M, Fujimura L, Yoshino H et al. Caveolin-1 mediates tumor cell migration and invasion and its regulation by miR-133a in head and neck squamous cell carcinoma. Int J Oncol 2011; 38: 209–217.
Lin DH, Yue P, Pan C, Sun P, Wang WH . MicroRNA 802 stimulates ROMK channels by suppressing caveolin-1. J Am Soc Nephrol 2011; 22: 1087–1098.
Zhang Y, Qu X, Li C, Fan Y, Che X, Wang X et al. miR-103/107 modulates multidrug resistance in human gastric carcinoma by downregulating Cav-1. Tumour Biol 2014; 36: 2277–2285.
Lino Cardenas CL, Henaoui IS, Courcot E, Roderburg C, Cauffiez C, Aubert S et al. miR-199a-5p Is upregulated during fibrogenic response to tissue injury and mediates TGFbeta-induced lung fibroblast activation by targeting caveolin-1. PLoS Genet 2013; 9: e1003291.
Ørom UA, Lim MK, Savage JE, Jin L, Saleh AD, Lisanti MP et al. MicroRNA-203 regulates caveolin-1 in breast tissue during caloric restriction. Cell Cycle 2012; 11: 1291–1295.
Chung WK, Sampson KS, Kass RS . A novel channelopathy in pulmonary arterial hypertension. N Engl J Med 2013; 369: 2162.
Girerd B, Perros F, Antigny F, Humbert M, Montani D . KCNK3: new gene target for pulmonary hypertension? Expert Rev Respir Med 2014; 8: 385–387.
Latus H, Delhaas T, Schranz D, Apitz C . Treatment of pulmonary arterial hypertension in children. Nat Rev Cardiol 2015; 12: 244–254.
Zamanian RT, Kudelko KT, Sung YK, de Jesus Perez V, Liu J, Spiekerkoetter E . Current clinical management of pulmonary arterial hypertension. Circ Res 2014; 115: 131–147.
Chester AH, Yacoub MH . The role of endothelin-1 in pulmonary arterial hypertension. Glob Cardiol Sci Pract 2014; 2014: 62–78.
Miyagawa K, Emoto N . Current state of endothelin receptor antagonism in hypertension and pulmonary hypertension. Ther Adv Cardiovasc Dis 2014; 8: 202–216.
Lai YC, Potoka KC, Champion HC, Mora AL, Gladwin MT . Pulmonary arterial hypertension: the clinical syndrome. Circ Res 2014; 115: 115–130.
Das A, Durrant D, Salloum FN, Xi L, Kukreja RC . PDE5 inhibitors as therapeutics for heart disease, diabetes and cancer. Pharmacol Ther 2015; 147C: 12–21.
Pabani S, Mousa SA .. Current and future treatment of pulmonary hypertension. Drugs Today (Barc) 2012; 48: 133–147.
Chrysant SG . Effectiveness and safety of phosphodiesterase 5 inhibitors in patients with cardiovascular disease and hypertension. Curr Hypertens Rep 2013; 15: 475–483.
Montani D, Chaumais MC, Guignabert C, Günther S, Girerd B, Jaïs X et al. Targeted therapies in pulmonary arterial hypertension. Pharmacol Ther 2014; 141: 172–191.
Tu L, Guignabert C . Emerging molecular targets for anti-proliferative strategies in pulmonary arterial hypertension. Handb Exp Pharmacol 2013; 218: 409–436.
Cavasin MA, Demos-Davies K, Horn TR, Walker LA, Lemon DD, Birdsey N et al. Selective class I histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ Res 2012; 110: 739–748.
Bogaard HJ, Mizuno S, Hussaini AA, Toldo S, Abbate A, Kraskauskas D et al. Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats. Am J Respir Crit Care Med 2011; 183: 1402–1410.
Esteller M, Fraga MF, Paz MF, Campo E, Colomer D, Novo FJ et al. Cancer epigenetics and methylation. Science 2002; 297: 1807–1808 discussion-8.
Schleithoff C, Voelter-Mahlknecht S, Dahmke IN, Mahlknecht U . On the epigenetics of vascular regulation and disease. Clin Epigenetics 2012; 4: 7.
Brock M, Haider TJ, Vogel J, Gassmann M, Speich R, Trenkmann M et al. The hypoxia-induced microRNA-130a controls pulmonary smooth muscle cell proliferation by directly targeting CDKN1A. Int J Biochem Cell Biol 2015; 61: 129–137.
Chen T, Zhou G, Zhou Q, Tang H, Ibe JC, Cheng H et al. Loss of microRNA-17∼92 in smooth muscle cells attenuates experimental pulmonary hypertension via induction of PDZ and LIM domain 5. Am J Respir Crit Care Med 2015; 191: 678–692.
Pullamsetti SS, Doebele C, Fischer A, Savai R, Kojonazarov B, Dahal BK et al. Inhibition of microRNA-17 improves lung and heart function in experimental pulmonary hypertension. Am J Respir Crit Care Med 2012; 185: 409–419.
Bertero T, Cottrill K, Krauszman A, Lu Y, Annis S, Hale A et al. The microRNA-130/301 family controls vasoconstriction in pulmonary hypertension. J Biol Chem 2015; 290: 2069–2085.
Bertero T, Lu Y, Annis S, Hale A, Bhat B, Saggar R et al. Systems-level regulation of microRNA networks by miR-130/301 promotes pulmonary hypertension. J Clin Invest 2014; 124: 3514–3528.
Jin Y, Pang T, Nelin LD, Wang W, Wang Y, Yan J et al. MKP-1 is a target of miR-210 and mediate the negative regulation of miR-210 inhibitor on hypoxic hPASMC proliferation. Cell Biol Int 2015; 39: 113–120.
Gou D, Ramchandran R, Peng X, Yao L, Kang K, Sarkar J et al. miR-210 has an antiapoptotic effect in pulmonary artery smooth muscle cells during hypoxia. Am J Physiol Lung Cell Mol Physiol 2012; 303: L682–L691.
Grant JS, Morecroft I, Dempsie Y, van Rooij E, MacLean MR, Baker AH . Transient but not genetic loss of miR-451 is protective in the development of pulmonary arterial hypertension. Pulm Circ 2013; 3: 840–850.
Sharma S, Umar S, Potus F, Iorga A, Wong G, Meriwether D et al. Apolipoprotein A-I mimetic peptide 4F rescues pulmonary hypertension by inducing microRNA-193-3p. Circulation 2014; 130: 776–785.
Shan F, Li J, Huang QY . HIF-1 alpha-induced up-regulation of miR-9 contributes to phenotypic modulation in pulmonary artery smooth muscle cells during hypoxia. J Cell Physiol 2014; 229: 1511–1520.
Li SS, Ran YJ, Zhang DD, Li SZ, Zhu D . MicroRNA-190 regulates hypoxic pulmonary vasoconstriction by targeting a voltage-gated K+ channel in arterial smooth muscle cells. J Cell Biochem 2014; 115: 1196–1205.
Kang BY, Park KK, Green DE, Bijli KM, Searles CD, Sutliff RL et al. Hypoxia mediates mutual repression between microRNA-27a and PPARγ in the pulmonary vasculature. PLoS ONE 2013; 8: e79503.
Kang K, Peng X, Zhang X, Wang Y, Zhang L, Gao L et al. MicroRNA-124 suppresses the transactivation of nuclear factor of activated T cells by targeting multiple genes and inhibits the proliferation of pulmonary artery smooth muscle cells. J Biol Chem 2013; 288: 25414–25427.
Li S, Ran Y, Zhang D, Chen J, Zhu D . MicroRNA-138 plays a role in hypoxic pulmonary vascular remodelling by targeting Mst1. Biochem J 2013; 452: 281–291.
Jalali S, Ramanathan GK, Parthasarathy PT, Aljubran S, Galam L, Yunus A et al. Mir-206 regulates pulmonary artery smooth muscle cell proliferation and differentiation. PLoS ONE 2012; 7: e46808.
Caruso P, Dempsie Y, Stevens HC, McDonald RA, Long L, Lu R et al. A role for miR-145 in pulmonary arterial hypertension: evidence from mouse models and patient samples. Circ Res 2012; 111: 290–300.
Guo L, Qiu Z, Wei L, Yu X, Gao X, Jiang S et al. The microRNA-328 regulates hypoxic pulmonary hypertension by targeting at insulin growth factor 1 receptor and L-type calcium channel-α1C. Hypertension 2012; 59: 1006–1013.
Courboulin A, Paulin R, Giguère NJ, Saksouk N, Perreault T, Meloche J et al. Role for miR-204 in human pulmonary arterial hypertension. J Exp Med 2011; 208: 535–548.
Acknowledgements
This study was supported by the Sookmyung Women’s University Research Grants of 1-1403-0050.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kim, JD., Lee, A., Choi, J. et al. Epigenetic modulation as a therapeutic approach for pulmonary arterial hypertension. Exp Mol Med 47, e175 (2015). https://doi.org/10.1038/emm.2015.45
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2015.45
This article is cited by
-
Unraveling the epigenetic landscape of pulmonary arterial hypertension: implications for personalized medicine development
Journal of Translational Medicine (2023)
-
Genome-wide identification, expression analysis, and functional verification of the JMJ (Jumonji) histone demethylase gene family in pear (Pyrus bretchneideri)
Tree Genetics & Genomes (2023)
-
Non-coding RNAs in human health and disease: potential function as biomarkers and therapeutic targets
Functional & Integrative Genomics (2023)
-
Isoform-specific characterization of class I histone deacetylases and their therapeutic modulation in pulmonary hypertension
Scientific Reports (2020)
-
Revealing the pathogenic changes of PAH based on multiomics characteristics
Journal of Translational Medicine (2019)
