
Abstract
The worldwide prevalence of obesity is steadily increasing, nearly doubling between 1980 and 2008. Obesity is often associated with insulin resistance, a major risk factor for type 2 diabetes mellitus (T2DM): a costly chronic disease and serious public health problem. The underlying cause of T2DM is a failure of the beta cells of the pancreas to continue to produce enough insulin to counteract insulin resistance. Most current T2DM therapeutics do not prevent continued loss of insulin secretion capacity, and those that do have the potential to preserve beta cell mass and function are not effective in all patients. Therefore, developing new methods for preventing and treating obesity and T2DM is very timely and of great significance. There is now considerable literature demonstrating a link between inhibitory guanine nucleotide-binding protein (G protein) and G protein-coupled receptor (GPCR) signaling in insulin-responsive tissues and the pathogenesis of obesity and T2DM. These studies are suggesting new and emerging therapeutic targets for these conditions. In this review, we will discuss inhibitory G proteins and GPCRs that have primary actions in the beta cell and other peripheral sites as therapeutic targets for obesity and T2DM, improving satiety, insulin resistance and/or beta cell biology.
Similar content being viewed by others
Introduction
Insulin resistance is a classic risk factor for the development of type 2 diabetes mellitus (T2DM).1, 2 A significant predisposition to insulin resistance is observed with obesity,3 classified as a body mass index greater than 30. Obesity and its associated comorbidities are reaching never-seen-before levels worldwide.4, 5 With sufficient increases in basal and nutrient-stimulated insulin levels, insulin resistance may not progress to T2DM.6 Systemic insulin levels can be increased by one of two general mechanisms: increased insulin secretion from individual beta cells (that is, beta cell function) and increased total numbers of beta cells (that is, beta cell mass).7
Reduced beta cell function in T2DM is clearly an early and important aspect underlying the pathophysiology of T2DM;8, 9, 10 hence, addressing this dysfunction is a valid therapeutic strategy for T2DM. (Little is known of the effects of the therapeutics discussed in this review on alpha cell function. As dysregulated glucagon secretion has also been noted as a key issue in T2DM pathophysiology, these are clearly important questions to address, but are outside of the scope of this review.) The first T2DM therapeutics to target the beta cells were the sulfonylurea compounds, which act directly on the ATP-sensitive potassium channels of the beta cell to enhance membrane depolarization, stimulating exocytosis of insulin granules.11 While still used clinically and excellent drugs in some circumstances, sulfonylureas act in a glucose-independent manner, increasing the potential risk of hypoglycemia.12 Furthermore, sulfonylureas and other long-established therapeutics, including the insulin-sensitizer metformin, do not prevent the continuing loss of beta cell function observed in T2DM.10 However, weight loss from metformin may indirectly benefit beta cell function and limit beta cell loss in T2DM.13, 14
A different aspect of beta cell dysfunction is being addressed by recent T2DM therapeutics: the lack of potentiation of insulin secretion in response to an oral glucose/nutrient challenge.15 This potentiation occurs because of peptide hormones termed incretins that are secreted from the digestive tract and act on the pancreas and other peripheral tissues to elicit biological effects, including augmenting nutrient-stimulated insulin secretion. This effect of incretins is lost or blunted in T2DM.16
The incretin receptors belong to a superfamily of transmembrane proteins termed as G protein-coupled receptors (GPCRs) that promote intracellular signal transduction pathways mediated by guanine nucleotide-binding proteins (G proteins). There are four major classes of G proteins: Gs, Gi, Gq, and G12.17 These proteins are classified based on the sequence identity of their alpha subunit, which confers similar downstream signaling mechanisms among subfamily members. Gs is the sole member of its subfamily not restricted to olfactory/taste sensory cells, while similar members of the Gi subfamily are Gi1, Gi2, Gi3, Go1, Go2 and Gz. The Gs and Gi subfamilies also include specialized members: Golf is a Gs subfamily member restricted to olfactory cells, while Gt and Ggust are Gi subfamily members restricted to the retina and taste cells, respectively. While taste and olfactory cell signaling is an interesting focus of current obesity and diabetes research (for example, see work from Dotson et al.18), only the more ubiquitously expressed G proteins will be discussed in this review.
The Gs and Gi subfamily members all signal by modulating the levels of the intracellular second-messenger cyclic AMP (cAMP), either stimulating (Gs) or inhibiting (Gi) the production of cAMP by adenylate cyclase. The impact of these proteins on cAMP production mirrors their effect on insulin secretion. For instance, most of the incretin receptors signal through Gs.19 cAMP has also been shown to regulate beta cell proliferation and survival, potentially linking these receptors to regulation of beta cell mass.20, 21 The Gq class of G proteins is also stimulatory toward insulin secretion, but acts through separate second-messenger pathways: inositol trisphosphate and diacylglycerol, which are formed when Gq-activated phospholipase C-β cleaves phosphatidylinositol bisphosphate.22 A smaller number of incretin receptors have been linked with Gq.23 Not as much is known regarding the role of G12 on beta cell function, but its actions through the small G protein, Rho, appear to be important for promoting the exocytosis process itself.24
Drugs targeting the glucagon-like peptide 1 (GLP-1) receptor, a Gs-coupled incretin receptor, are some of the newer T2DM therapeutics, having been used clinically for nearly a decade.25 The stable GLP-1 analogs, such as exenatide (also known as Byetta, Amylin Pharmaceuticals, San Diego, CA, USA), are resistant to inactivating cleavage by dipeptidyl peptidase 4, which rapidly degrades endogenous GLP-1. In addition, endogenous GLP-1 can be stabilized by treatment with dipeptidyl peptidase inhibitors, such as sitagliptin (also known as Januvia, Merck, Whitehouse Station, NJ, USA). Several other Gq- and Gs-coupled incretin receptors are being explored as T2DM therapeutic targets and have been reviewed elsewhere.21, 26, 27 Even with these successes, incretin-based therapeutics do not work in all individuals and safety concerns, including pancreatitis and gastrointestinal distress, in addition to possible side effects such as weight changes lead to non-compliance.28 These concerns raise the possibility of targeting potential dysfunction in other components of beta cell signaling pathways.
Endogenous inhibitors of beta cell function have long been known, even before the identity of their receptors and associated G proteins were established. For example, alpha adrenergic receptor (α-AR) agonists such as epinephrine and norepinephrine are released from the sympathetic nervous system as part of the fight-or-flight response, one aspect of which is to maintain higher glucose levels in the bloodstream.29 α-ARs are expressed in beta cells, and agonists of the α2-AR subfamily, which are Gi-coupled receptors, rapidly reduce glucose-stimulated insulin secretion.30 Another long-established inhibitor of beta cell function, prostaglandin E2 (PGE2), is produced by cyclooxygenase (COX)-catalyzed metabolism of arachidonic acid and acts in an autocrine and/or paracrine fashion to modulate beta cell growth and insulin secretion.31 Interestingly, receptors for adrenergic agonists and PGE2, as well as their associated G proteins, have all been linked with diabetic beta cell pathophysiology.32, 33, 34, 35
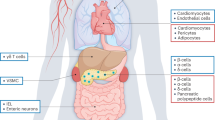
Insulin-responsive tissues besides the pancreas also play critical roles in the pathophysiology of T2DM. The pancreatic circulation empties directly into the portal vein, meaning that the liver is the first stop for hormones secreted from the pancreas, including insulin and glucagon. The liver is critical for the maintenance of whole-body glucose homeostasis. Insulin acts on the liver to signal the fed state of the body, promoting glucose storage into glycogen and blocking gluconeogenesis and lipolysis, blocking hepatic glucose and free fatty acid production. It is well accepted that hepatic fuel production is dysfunctional in T2DM, as insulin-resistant alpha cells secrete glucagon in an unregulated manner, further exacerbating the phenotype of liver insulin resistance.36, 37 In addition, muscle and adipose tissue are stimulated by insulin to increase glucose uptake for use as fuel and storage as glycogen and triglycerides, respectively. Inefficient muscle and adipose glucose uptake is also observed in T2DM and contributes to the pathology of hyperglycemia.38, 39 There is much debate surrounding the impact of enhanced beta cell function and mass to the development of insulin resistance in T2DM. This discussion is beyond the scope of this review, but has been well described elsewhere.40 Specific GPCRs, including melatonin receptor isoform 2 (MT2), have been shown to have important roles in regulating insulin sensitivity.41
Metabolic syndrome is often linked to obesity and visceral fat accumulation known to perturb blood glucose regulation and increase insulin resistance; however, obesity by itself is not a definitive predictor of metabolic syndrome as some obese individuals do not present with typical etiologies.42 A diagnosis of metabolic syndrome is a significant risk factor, though, for the development of T2DM, cardiovascular disease, neurodegeneration, polycystic ovarian syndrome, cancer and non-alcoholic fatty liver disease, among others.43 Therefore, developing anti-obesity therapeutics as a preventative for these significant, chronic diseases is a major focus of current research. Several effective medical treatments for obesity have been removed from the market or are strictly regulated due to unacceptable side effects. Fenfluramine/phentermine (also known as phen-fen) was pulled from the US market in 1997. Sibutramine (also known as Reductil, Meridia and Sibutrex, Abbott Labs, Abbott Park, IL, USA) is a schedule IV-controlled substance and was pulled from the US market in 2010. Dextromethamphetamine (also known as Desoxyn, Abbot Labs) is a methamphetamine derivative and a schedule II-controlled substance. Furthermore, the few widely available medical treatments such as orlistat (also known as Xenical, Roche, Basel, Switzerland; and Alli, GlaxoSmithKline, Research Triangle Park, NC, USA) have only modest effects at best.44 Surgical techniques such as gastric bypass and sleeve gastrectomy are often quite effective, but are also major procedures that have continuously decreasing, yet still not insignificant, peri- and post-operative morbidity and mortality risks.45 A safe, effective medical treatment to augment healthy lifestyle changes in the efforts to combat obesity and metabolic syndrome is sorely needed. One inhibitory GPCR in particular, cannabinoid receptor type 1 (CB1) has been demonstrated as a potentially effective anti-obesity pharmacological target.
In this review, we will discuss Gi subfamily members that have been linked to beta cell dysfunction, insulin resistance or both. Next, we will discuss a select number of inhibitory GPCRs that have been shown to have significant pre-clinical or clinical evidence of efficacy as anti-T2DM and/or anti-obesity targets. These include GPCRs whose major effect is in the beta cell, as potential targets to maintain beta cell function and/or mass; and GPCRs with primary effects in other insulin-responsive tissues, as targets to improve insulin resistance and/or obesity. It is our hope that this review will stimulate further research into these relatively under-studied proteins as potentially useful therapeutic targets for the obesity and T2DM that is becoming a worldwide public health problem.
Gi subfamily proteins as targets for beta cell dysfunction in T2DM
Beta cell dysfunction and insulin resistance are early and necessary events in the development of T2DM,8 and Gi signaling has been shown to be an important regulator of both these events. The Gi family inhibitor, pertussis toxin (PTX), was originally named islet-activating protein due to its ability to promote glucose- and hormone-stimulated insulin secretion long before its mechanism of action was known.46 Even so, no insulin secretion phenotypes had been reported in mouse lines deficient in individual Gi subfamily members, until those lacking only the PTX-insensitive Gi subfamily alpha subunit, Gαz, were studied (Since then, other Gi subfamily members that are sensitive to PTX have been linked with regulating beta cell function in vivo, explaining the effect of PTX on insulin secretion: these will be discussed in more detail below). By sequence and functional similarity, Gz is clearly a member of the Gi subfamily, but it is unique in that its alpha subunit (Gαz) lacks the C-terminal cysteine residue that is the substrate for PTX-mediated ADP ribosylation and inactivation.47 Gαz-null mice exhibit improved insulin secretion response and glucose clearance after glucose challenge, correlating with constitutively increased cAMP levels and improved glucose-stimulated insulin secretion from their isolated islets.48 These effects were observed even though no GPCR had been specifically targeted. The slow Gαz GTP hydrolysis and deactivation rate were posited as a potential explanation for constitutive effects,47, 48 although endogenous beta cell GPCRs have been linked specifically with Gz. Agonists activating the prostaglandin E receptor 3 (EP3) isoform of the PGE2 receptor block glucose-stimulated insulin secretion from insulinoma cells and isolated mouse islets in a PTX-insensitive manner, suggesting coupling of the EP3 receptor specifically to Gz and not other Gi proteins.33, 49 These results were confirmed with specific inhibitors of Gαz itself.49 In addition, the modulation of endocytosis following exocytosis (a required event in the maintenance of insulin secretion) by norepinephrine is also PTX-insensitive. A synthetic peptide mimicking the C-terminus of the Gαz blocked this norepinephrine effect.50
There appears to be no effect of Gαz on insulin sensitivity. This is not unexpected, as Gαz has little-to-no expression in the liver, adipose tissue or skeletal muscle,47, 51, 52 and although expressed in the brain, does not seem to regulate appetite or energy metabolism.33 Even so, Gαz-null mice are completely protected from the development of glucose intolerance upon high-fat diet feeding, primarily through a significant augmentation of beta cell mass and maintenance of beta cell function.33 These results suggest Gαz as an emerging target to improve both defects of the diabetic islet: reduced beta cell mass and decreased beta cell function.
Other Gi subfamily members, including both Gi and Go isoforms, have been implicated in directly inhibiting exocytosis from beta cells,53 which, together with their subcellular localization, indicates a role for Gi proteins in the regulation of a distal step in the stimulated secretion pathway. In studies in an insulinoma cell line, norepinephrine inhibited the refilling of the readily releasable pool of secretory granules, an effect that was abolished by a dual Gαi1/2-blocking peptide.54 Perplexingly, mice deficient in Gαi2 expression did not have noticeably improved insulin secretion, but instead had reduced insulin sensitivity.55 Furthermore, expression of constitutively active Gαi2 in mouse liver and adipose tissue enhanced glucose tolerance.56 Among other effects, a direct effect of Gαi2 signaling on GLUT4 membrane translocation through a phosphoinositide 3-kinase-dependent mechanism may be responsible for these phenotypes.57
Early studies with Gαo-null mice indicated changes in the nervous system structure and aberrant heart calcium channel signaling, but no effect on blood glucose levels.58 In more recent work, one of the two splice variants of Gαo, Gαo2, was revealed as a key regulator of insulin secretion. Mice lacking Gαo2, but not those lacking Gαo1 or any Gαi isoforms have augmented glucose-stimulated insulin secretion and improved glucose tolerance,59 just as observed with Gαz-null mice. Moreover, these results help support earlier findings that targeting PTX-sensitive proteins, such as Gαo2, can potentiate insulin secretion but in a different manner than Gαz.46, 49 These results are important, as they suggest that the Gi subfamily members in the beta cell are not redundant, as has been suggested in other systems60 and that specific isoforms and/or their associated receptors might be useful T2DM therapeutic targets. Of note, no small-molecule pharmaceutical agents in use clinically target a GPCR/G protein pathway directly at the G protein level (although some have been proposed hypothetically and/or tested in vitro61, 62, 63). In addition, biased GPCR ligands can preferentially effect one versus another G protein-dependent or -independent downstream signaling pathway, but these are still receptor-based drugs and not G protein-based.64 Thus, understanding more about the endogenous GPCRs that these inhibitory G proteins couple to will likely be very important in pharmaceutically targeting their activation.
The EP3 receptor: targeting dysfunctional agonist production and receptor signaling in T2DM
The pathophysiology of T2DM is increasingly being linked with inflammation.65, 66, 67 Many metabolites of the ω-6 polyunsaturated fatty acid (PUFA) arachidonic acid have been linked directly with inflammation.68, 69 E-series prostaglandins, comprising a major family of arachidonate metabolites, were posited as playing a role in T2D over 30 years ago.70, 71 PGE2 in particular has long been known as a physiological inhibitor of insulin secretion, acting via autocrine or paracrine mechanisms.72 This means that islet cells themselves, including beta cells, express the enzymes required for synthesis of PGE2, including phospholipase A2, which cleaves arachidonate from plasma membrane phospholipids; COX, which convert the fatty acid into the intermediate PGH2; and PGE synthases (Ptges), which convert PGH2 to PGE2.
The ω-3 PUFAs compete with ω-6 PUFAs for some of the same metabolic enzymes. Many groups (including the American Diabetes Association) have recommended a high ω-3 PUFA diet as a means to prevent disease.73 Yet, there still exists some controversy as to the benefits of ω-3 PUFAs on chronic disease74, 75 as well as the impact of high ω-3 PUFAs in the context of a diet that remains high in ω-6 PUFAs. Furthermore, few studies have explored these interventions specifically in the beta cell. Notable exceptions include two groups that generated transgenic mice expressing a Caenorhabditis elegans desaturase that converts ω-6 PUFAs to the ω-3 form. These mice robustly increased ω-3 PUFA concentrations (and reduced ω-6 PUFAs to near zero) in all tissues, with improved insulin secretion from isolated islets.76, 77
Inhibition of COX (which, in beta cells, is primarily COX-231, 78) can completely block PGE2 production along with that of numerous other metabolites downstream of PGH2 and the other COX intermediates. The upregulation of COX-2 expression and/or activity has been clearly linked with the diabetic state.66, 79, 80, 81, 82 The potential utility of COX-2 inhibitors in T2DM is overshadowed by concerns of significant cardiovascular risks, especially in a population at greater intrinsic risk of cardiovascular disease.83, 84 Interestingly, after nearly a half century, there has been a recent resurgence in studies on the utility of salicylate, which acts directly at the site of inflammation to reduce COX-1 and COX-2 expression, as a T2D therapeutic.85, 86
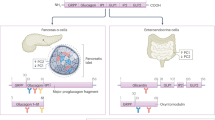
Recently, a specific PGE2 receptor isoform, EP3, has been suggested as an emerging target for T2DM therapeutics. EP3 receptor expression and activity was significantly upregulated in diabetic mouse islets, as was the production of PGE2.32 Targeting the EP3 receptor with a specific antagonist, L798,106, significantly improved the insulin secretory response of diabetic mouse and human islets.32 Interestingly, EP3 signaling was found to oppose the action of the GLP-1 receptor, discussed above as a significant potentiator of glucose-stimulated insulin secretion. Specifically, the maximal effect of GLP-1 on insulin secretion was blunted by EP3 activation, and a combination of an EP3 antagonist and GLP-1 agonist additively promoted insulin secretion from diabetic mouse islets.32 The mechanisms mediating dysfunctional EP3 signaling in diabetic beta cells and potential ways to target EP3 in T2DM therapy are shown on the left half of Figures 1.

Summary of some of the proposed mechanisms of dysfunctional E prostanoid receptor 3 (EP3) and α2A-adrenergic receptor (α2A-AR) signaling in diabetic beta cells and how these receptors might be targeted in type 2 diabetes mellitus (T2DM) therapy. In a schematic of a diabetic beta cell, both expression and/or activity of both EP3 (left) and α2A-AR (right) have been shown to be dysfunctionally upregulated. Increased EP3 expression/activity is exacerbated by increased prostaglandin E2 (PGE2) production, acting in an autocrine/paracrine manner to further reduce cyclic AMP (cAMP) production by Gi subfamily member, Gz, signaling to adenylate cyclase (AC). In contrast, the release of α2A-AR agonists epinephrine and norepinephrine (Epi/NE) after stimulation by the parasympathetic nervous system (PNS) is not necessarily dysfunctionally upregulated in T2DM; rather, a specific ADRA2A single nucleotide polymorphism (SNP) confers increased stability of α2A-AR at the plasma membrane, allowing Epi/NE to tonically signal through α2A-AR and associated Gi subfamily proteins to reduce cAMP production by AC. Of note, cAMP is one of the only signaling pathways shown to positively impact on both beta cell mass (that is, growth, proliferation and survival) and beta cell function (that is, insulin secretion). Gray arrows and text indicate confirmed or potential downregulation of these effects with dysfunctional EP3 or α2A-AR signaling. The EP3 antagonist, L798,106, has been shown to reverse diabetic beta cell dysfunction in isolated islets in vitro, while DG-041 has gone through clinical trials for a different indication, indicating potential in vivo utility. Also suggested in this figure is the potential to target EP3 signaling by reducing PGE2 production with nutritional or pharmacological interventions. With regards to α2A-AR, a specific antagonist, yohimbine, improves insulin secretion from islets isolated from individuals with the specific ADRA2A SNP conferring α2A-AR stability. A clinical trial to confirm this finding in vivo has been initiated. Again, both EP3 and α2A-AR antagonists have the potential to improve beta cell mass, but this has yet to be confirmed.
Although the EP3 receptor plays critical roles in islet biology, extrapancreatic function may be equally important in T2DM pathophysiology; in particular, in mediating T2DM complications. The migratory response of vascular smooth muscle cells is required for the dysfunctional vascular remodeling observed with cardiovascular disease. Genetic deletion or pharmaceutical inhibition of EP3 results altered polarity and directional migration of vascular smooth muscle cells, suggesting that blockade of EP3 might protect from dysfunctional vascular remodeling.87 In addition, mice lacking EP3 have reduced baseline and stimulated mean arterial pressures, and pharmacological inhibition of EP3 blocks angiotensin 2-mediated vasoconstriction.88 Thus, it appears that PGE2 signaling through EP3 may play a significant role in the progression of T2DM and its associated comorbidities.
Mice lacking EP3 develop obesity, insulin resistance, glucose intolerance and eat considerably more than wild-type controls.89 Part of this phenotype can be explained by increased light cycle eating, most likely due to unstable sleep patterns, as PGE2 (acting through hypothalamic EP3) may act as a somnogen.90 Furthermore, high levels of PGE2 and increased signaling through EP3 both augment nitric oxide synthase expression, an enzyme critical in the brain development of newborns.91 This signaling cascade is suggested to play a significant role in connecting brain circulation and synaptic activity in perinatal development. If EP3 antagonists are pursued as therapeutics for T2DM and/or metabolic disease, a peripherally restricted EP3 antagonist might be necessary. Interestingly, an EP3 antagonist, DG-041, completed a European phase II clinical trial by Decode Genetics testing its effect in treating peripheral vascular disease, which is typically caused and/or complicated by hypertension. The results of this trial were never published, so questions remain as to whether this drug was effective as an anti-hypertensive agent. Recently, DG-041 treatment in mice was found to limit atherothrombosis without altering hemostasis, suggesting that DG-041 can decrease clot formation without increasing bleeding risk, critical in the prevention of cardiovascular disease.92
The α2A AR: targeting genetic susceptibility to T2DM
Dysfunctional α-AR signaling was noted in T2DM at least 40 years ago.34, 93 In an early clinical study of six diabetic individuals, α-AR blockade with phentolamine restored the weak diabetic insulin secretion response to glucose.34 α-AR antagonists were never much pursued as anti-diabetic agents, though, perhaps due to the systemic effects of α-AR signaling, including smooth muscle contraction, inhibition of neurotransmitter release, transient hypertension followed by sustained hypotension, and constriction of certain veins and arteries, including those leading to and from the heart, among others.
This possible thinking that α-AR signaling was too pleiotropic to be targeted therapeutically in T2DM changed in 2010, when a single nucleotide polymorphism in the α2A-AR gene, ADRA2A, was discovered as a susceptibility factor for T2D: rs553668.35 In two different studies, including over 5000 individuals, Rosengren et al.35, 94 showed that individuals with a specific rs553668 genotype trended toward elevated fasting plasma glucose levels and reduced serum insulin levels. Furthermore, these individuals were significantly more prone to a hypoinsulinemic response during glucose tolerance testing, indicating a defect in the insulin secretion process itself.35, 94 When human pancreatic islet preparations were genotyped, those with the specific rs553668 variant had reduced insulin granule docking and secreted less insulin in response to glucose due to increased expression of α2A-AR at the membrane.35, 94 Treatment of isolated islets with an α2A-AR antagonist, yohimbine, corrected the insulin secretory dysfunction in these islets.35 A clinical trial initiated in the US is currently enrolling patients with this specific genotypic risk variant in a randomized, controlled trial of yohimbine as a T2D therapeutic.
A more recent meta-analysis of four prospective studies confirmed the linkage of rs553668 with fasting glucose and T2DM risk, and found another variant of ADRA2A, rs17186196, associated with fasting glucose levels, fasting insulin levels and whole-body insulin sensitivity.95 Furthermore, rs491589 was associated with systolic and diastolic blood pressure, while rs36022820 correlated with body mass index.95 Additional ADRA2A variants have been linked with hypertension, a significant risk factor for cardiovascular disease, a common complication of T2DM.96, 97 In a study of 70 hypertensive patients, 35 with and 35 without T2DM, individuals with a genotype encoding for a small deletion in ADRA2A were significantly more likely to have hypertension, regardless of T2DM status.98 A potential physiological mechanism for this association was the significantly lower high-density lipoprotein and higher low-density lipoprotein in individuals homozygous for the deletion phenotype.98 Overall, these results confirm the utility of targeting the α2A-AR to improve beta cell function in T2DM in a specific population, and also indicate its possible utility as a target for insulin resistance, obesity and/or T2DM complications. The mechanisms mediating dysfunctional α2A-AR activity in a specific subset of diabetic beta cells and potential ways to target α2A-AR in T2DM therapy are shown on the right half of Figure 1.
The MT2 receptor: targeting dysregulated insulin secretion and peripheral insulin resistance
It has been demonstrated in humans that disruption of the circadian rhythm promotes obesity and diabetes, and leads to perturbations in plasma glucose and insulin concentrations.99 For instance, melatonin (N-acetyl-5-methoxytryptamine) is secreted from the pineal gland and has a major role in the regulation of circadian rhythms. It has also been shown to affect carbohydrate metabolism and insulin secretion.100 Melatonin acts through two receptors, MT1 (gene name: MTNR1A) and MT2 (gene name: MTNR1B). Activation of MT1 or MT2 receptors in the beta cell inhibit insulin secretion.100 However, it is not melatonin antagonists that have been proposed as obesity/T2DM therapeutics, but rather melatonin agonists. This is because MT1/MT2 receptor signaling also reduces dyslipidemia, reduces hepatic glucose output, decreases adiposity and improves metabolic function.41, 101, 102 Low melatonin levels have also been independently associated with T2DM risk.103 Thus, stimulating melatonin signaling has been proposed as a treatment for metabolic syndrome.41
Altered melatonin receptor expression or loss-of-function mutations and altered melatonin secretion itself have all been previously linked with T2DM or glucose intolerance.103 Melatonin was shown to function by reducing hepatic gluconeogenesis and phosphorylation and activation of Akt in the hypothalamus.101 Most of the recent research into the links between the melatonin receptors and diabetes is focused on MT2. Independent genome-wide association studies in European populations identified single nucleotide polymorphisms near MTNR1B that were associated with fasting plasma glucose, reduced beta cell function, and significantly increased T2DM risk: rs1387153 and rs10830963.104, 105 Expression of these two single nucleotide polymorphisms is also significantly associated with the development of gestational diabetes,106, 107 as well as elevated fasting glucose and reduced insulin secretion after parturition.106 A diagnosis of gestational diabetes is one of the highest predictors for the later development of T2DM.106 A clinical trial was registered in late 2012 to test the effects of 3 months of melatonin treatment on glucose tolerance in individuals with different MTNR1B variants.
The CB1 receptor: targeting appetite and satiety as an anti-obesity therapeutic
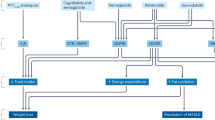
CB1 is a Gi-coupled receptor located in the central and peripheral nervous system as well as other insulin-responsive tissues. Endogenous CB1 ligands include anandamide (which is responsible for pleasurable effects, such as that from eating chocolate) and 2-arachidonoyl glyceride.108 Natural and synthetic cannabinoids such as tetrahydrocannabinol, the main active compound of the psychoactive drug cannabis, and its synthetic analog, dronabinol, are potent CB1 agonists. In the brain, CB1 is highly expressed in the hypothalamus, which regulates appetite, and the medulla, which contains the chemoreceptor trigger zone, regulating nausea and vomiting, and where it is involved in the regulation of normal brain aging.108 This explains why tetrahydrocannabinol is such an effective drug at combatting the nausea and loss of appetite associated with chemotherapy. Despite potential side effects such as nausea, the appetite-suppressive effects of central CB1 stimulated research into antagonists of the receptor as potential anti-obesity therapeutics.44
Several trials of the systemic CB1 antagonist, rimonabant, as a weight-loss agent were initiated in both Europe and North America. Overall, rimonabant was superior to both sibutramine and orlistat, with a nearly 5-kg average weight loss over a 1-year period (vs 4.5 kg for sibutramine and 2.5 kg for orlistat).44 Also, T2DM patients treated with rimonabant in addition to metformin or sulfonylurea had significantly lower HbA1c levels than those treated with metformin or sulfonylurea alone.109 This effect was at least partially independent of changes in body mass index. Not surprisingly, nausea was a common adverse effect in the rimonabant group (13%, as compared with 4% with placebo), as well as were a number of severe psychological alterations. Specifically, 15% percent of rimonabant-treated patients withdrew from the trials, and nearly half of these dropouts were attributed to mood alterations such as depression and increased suicide ideation.109 These outcomes halted any ongoing rimonabant trials, and the CB1 antagonist field made little progress over the next few years.
Perhaps due to the importance of CB1 in mediating appetite in the central nervous system, alternative CB1 antagonists had not been tested until recently. These include the neutral CB1 antagonists, which have a weaker affinity for receptor, but also peripherally restricted CB1 antagonists, which cannot cross the blood-brain barrier and thus have no direct central nervous system effects. Insulin-responsive tissues such as the liver, pancreas, adipose and muscle also express CB1, and antagonism of these receptors promotes insulin signaling, blocks lipogenesis, promotes fatty acid oxidation and stimulates energy-requiring processes such as mitochondrial biogenesis and cell growth and differentiation.110 CB1 receptors are also located on afferent and efferent nerve fibers; thus, the insulin-responsive state of the body is transmitted to the brain, where it can send signals to decrease food intake.110 Overall, a requirement for direct central nervous system activation of CB1 receptors does not seem to exist in order to elicit positive metabolic changes, and perhaps, weight loss. Several recent publications show strong pre-clinical efficacy of these peripherally restricted CB1 antagonists.111, 112, 113
Summary
In recent years, we have learned much about the role of inhibitory G proteins and GPCRs in mediating dysfunctional signaling pathways in metabolic diseases such as T2DM and confirmed their efficacy in pre-clinical models and select human studies. A number of other inhibitory GPCRs not discussed above may also have therapeutic potential (for example, serotonin 2A, ghrelin, NPY and GPR41, among others; reviewed by Ahren26). Yet, we still have a long way to go before demonstrating that any of these proteins are suitable targets for T2DM and obesity in the general population. Even so, the current results are quite promising.
References
Haffner SM . Epidemiology of type 2 diabetes: risk factors. Diabetes Care 1998; 21 (Suppl 3): C3–C6.
DeFronzo RA . Pathogenesis of type 2 (non-insulin dependent) diabetes mellitus: a balanced overview. Diabetologia 1992; 35: 389–397.
Qatanani M, Lazar MA . Mechanisms of obesity-associated insulin resistance: many choices on the menu. Genes Dev 2007; 21: 1443–1455.
Shaw JE, Sicree RA, Zimmet PZ . Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract 2010; 87: 4–14.
Wild S, Roglic G, Green A, Sicree R, King H . Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 2004; 27: 1047–1053.
Gerich JE . Contributions of insulin-resistance and insulin-secretory defects to the pathogenesis of type 2 diabetes mellitus. Mayo Clin Proc 2003; 78: 447–456.
Meier JJ, Bonadonna RC . Role of reduced beta-cell mass versus impaired beta-cell function in the pathogenesis of type 2 diabetes. Diabetes Care 2013; 36 (Suppl 2): S113–S119.
Kahn SE . The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia 2003; 46: 3–19.
Kanat M, Mari A, Norton L, Winnier D, DeFronzo RA, Jenkinson C et al. Distinct beta-cell defects in impaired fasting glucose and impaired glucose tolerance. Diabetes 2012; 61: 447–453.
Defronzo RA . Banting lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009; 58: 773–795.
Doyle ME, Egan JM . Pharmacological agents that directly modulate insulin secretion. Pharmacol Rev 2003; 55: 105–131.
Srivastava S, Saxena GN, Keshwani P, Gupta R . Comparing the efficacy and safety profile of sitagliptin versus glimepiride in patients of type 2 diabetes mellitus inadequately controlled with metformin alone. J Assoc Physicians India 2012; 60: 27–30.
Glueck CJ, Fontaine RN, Wang P, Subbiah MT, Weber K, Illig E et al. Metformin reduces weight, centripetal obesity, insulin, leptin, and low-density lipoprotein cholesterol in nondiabetic, morbidly obese subjects with body mass index greater than 30. Metabolism 2001; 50: 856–861.
Lee A, Morley JE . Metformin decreases food consumption and induces weight loss in subjects with obesity with type II non-insulin-dependent diabetes. Obes Res 1998; 6: 47–53.
Nauck M, Stockmann F, Ebert R, Creutzfeldt W . Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 1986; 29: 46–52.
Bagger JI, Knop FK, Lund A, Vestergaard H, Holst JJ, Vilsboll T . Impaired regulation of the incretin effect in patients with type 2 diabetes. J Clin Endocrinol Metab 2011; 96: 737–745.
Strathmann M, Wilkie TM, Simon MI . Diversity of the G-protein family: sequences from five additional alpha subunits in the mouse. Proc Natl Acad Sci USA 1989; 86: 7407–7409.
Dotson CD, Vigues S, Steinle NI, Munger SD . T1R and T2R receptors: the modulation of incretin hormones and potential targets for the treatment of type 2 diabetes mellitus. Curr Opin Investig Drugs 2010; 11: 447–454.
Furman B, Pyne N, Flatt P, O'Harte F . Targeting beta-cell cyclic 3'5' adenosine monophosphate for the development of novel drugs for treating type 2 diabetes mellitus. A review. J Pharm Pharmacol 2004; 56: 1477–1492.
Baggio LL, Drucker DJ . Therapeutic approaches to preserve islet mass in type 2 diabetes. Annu Rev Med 2006; 57: 265–281.
Garber AJ . Incretin effects on beta-cell function, replication, and mass: the human perspective. Diabetes Care 2011; 34 (Suppl 2): S258–S263.
Sassmann A, Gier B, Grone HJ, Drews G, Offermanns S, Wettschureck N . The Gq/G11-mediated signaling pathway is critical for autocrine potentiation of insulin secretion in mice. J Clin Invest 2010; 120: 2184–2193.
Rehfeld JF . Incretin physiology beyond glucagon-like peptide 1 and glucose-dependent insulinotropic polypeptide: cholecystokinin and gastrin peptides. Acta Physiol (Oxf) 2011; 201: 405–411.
Wang Z, Thurmond DC . Mechanisms of biphasic insulin-granule exocytosis—roles of the cytoskeleton, small GTPases and SNARE proteins. J Cell Sci 2009; 122: 893–903.
Gallwitz B . New therapeutic strategies for the treatment of type 2 diabetes mellitus based on incretins. Rev Diabet Stud 2005; 2: 61–69.
Ahren B . Islet G protein-coupled receptors as potential targets for treatment of type 2 diabetes. Nat Rev Drug Discov 2009; 8: 369–385.
Winzell MS, Ahren B . G-protein-coupled receptors and islet function-implications for treatment of type 2 diabetes. Pharmacol Ther 2007; 116: 437–448.
Nisal K, Kela R, Khunti K, Davies MJ . Comparison of efficacy between incretin-based therapies for type 2 diabetes mellitus. BMC Med 2012; 10: 152.
Nonogaki K . New insights into sympathetic regulation of glucose and fat metabolism. Diabetologia 2000; 43: 533–549.
Hsu WH, Xiang HD, Rajan AS, Boyd AE 3rd . Activation of alpha 2-adrenergic receptors decreases Ca2+ influx to inhibit insulin secretion in a hamster beta-cell line: an action mediated by a guanosine triphosphate-binding protein. Endocrinology 1991; 128: 958–964.
Robertson RP . Dominance of cyclooxygenase-2 in the regulation of pancreatic islet prostaglandin synthesis. Diabetes 1998; 47: 1379–1383.
Kimple ME, Keller MP, Rabaglia MR, Pasker RL, Neuman JC, Truchan NA et al. Prostaglandin E2 receptor, EP3, is induced in diabetic islets and negatively regulates glucose- and hormone-stimulated insulin secretion. Diabetes 2013; 62: 1904–1912.
Kimple ME, Moss JB, Brar HK, Rosa TC, Truchan NA, Pasker RL et al. Deletion of Gαz protein protects against diet-induced glucose intolerance via expansion of β-cell mass. J Biol Chem 2012; 287: 20344–20355.
Robertson RP, Halter JB, Porte D Jr . A role for alpha-adrenergic receptors in abnormal insulin secretion in diabetes mellitus. J Clin Invest 1976; 57: 791–795.
Rosengren AH, Jokubka R, Tojjar D, Granhall C, Hansson O, Li DQ et al. Overexpression of alpha2A-adrenergic receptors contributes to type 2 diabetes. Science 2010; 327: 217–220.
Michael MD, Kulkarni RN, Postic C, Previs SF, Shulman GI, Magnuson MA et al. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell 2000; 6: 87–97.
Kawamori D, Kurpad AJ, Hu J, Liew CW, Shih JL, Ford EL et al. Insulin signaling in alpha cells modulates glucagon secretion in vivo. Cell Metab 2009; 9: 350–361.
Kim JK, Michael MD, Previs SF, Peroni OD, Mauvais-Jarvis F, Neschen S et al. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J Clin Invest 2000; 105: 1791–1797.
Guilherme A, Virbasius JV, Puri V, Czech MP . Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol 2008; 9: 367–377.
Gerich JE . The genetic basis of type 2 diabetes mellitus: impaired insulin secretion versus impaired insulin sensitivity. Endocr Rev 1998; 19: 491–503.
Srinivasan V, Ohta Y, Espino J, Pariente JA, Rodriguez AB, Mohamed M et al. Metabolic syndrome, its pathophysiology and the role of melatonin. Recent Pat Endocr Metab Immune Drug Discov 2013; 7: 11–25.
Despres JP, Lemieux I . Abdominal obesity and metabolic syndrome. Nature 2006; 444: 881–887.
Biddinger SB, Kahn CR . From mice to men: insights into the insulin resistance syndromes. Annu Rev Physiol 2006; 68: 123–158.
Gadde KM, Allison DB . Cannabinoid-1 receptor antagonist, rimonabant, for management of obesity and related risks. Circulation 2006; 114: 974–984.
Vetter ML, Dumon KR, Williams NN . Surgical treatments for obesity. Psychiatr Clin North Am 2011; 34: 881–893.
Katada T, Ui M . Islet-activating protein. Enhanced insulin secretion and cyclic AMP accumulation in pancreatic islets due to activation of native calcium ionophores. J Biol Chem 1979; 254: 469–479.
Casey PJ, Fong HK, Simon MI, Gilman AG . Gz, a guanine nucleotide-binding protein with unique biochemical properties. J Biol Chem 1990; 265: 2383–2390.
Kimple ME, Joseph JW, Bailey CL, Fueger PT, Hendry IA, Newgard CB et al. G{alpha}z negatively regulates insulin secretion and glucose clearance. J Biol Chem 2008; 283: 4560–4567.
Kimple ME, Nixon AB, Kelly P, Bailey CL, Young KH, Fields TA et al. A role for G(z) in pancreatic islet beta-cell biology. J Biol Chem 2005; 280: 31708–31713.
Zhao Y, Fang Q, Straub SG, Lindau M, Sharp GW . Hormonal inhibition of endocytosis: novel roles for noradrenaline and G protein G(z). J Physiol 2010; 588: 3499–3509.
Matsuoka M, Itoh H, Kozasa T, Kaziro Y . Sequence analysis of cDNA and genomic DNA for a putative pertussis toxin-insensitive guanine nucleotide-binding regulatory protein alpha subunit. Proc Natl Acad Sci USA 1988; 85: 5384–5388.
Garibay JL, Kozasa T, Itoh H, Tsukamoto T, Matsuoka M, Kaziro Y . Analysis by mRNA levels of the expression of six G protein alpha-subunit genes in mammalian cells and tissues. Biochim Biophys Acta 1991; 1094: 193–199.
Lang J . Molecular mechanisms and regulation of insulin exocytosis as a paradigm of endocrine secretion. Eur J Biochem 1999; 259: 3–17.
Zhao Y, Fang Q, Straub SG, Lindau M, Sharp GW . Noradrenaline inhibits exocytosis via the G protein betagamma subunit and refilling of the readily releasable granule pool via the alpha(i1/2) subunit. J Physiol 2010; 588: 3485–3498.
Moxham CM, Malbon CC . Insulin action impaired by deficiency of the G-protein subunit G ialpha2. Nature 1996; 379: 840–844.
Chen JF, Guo JH, Moxham CM, Wang HY, Malbon CC . Conditional, tissue-specific expression of Q205L G alpha i2 in vivo mimics insulin action. J Mol Med 1997; 75: 283–289.
Song X, Zheng X, Malbon CC, Wang H . Galpha i2 enhances in vivo activation of and insulin signaling to GLUT4. J Biol Chem 2001; 276: 34651–34658.
Valenzuela D, Han X, Mende U, Fankhauser C, Mashimo H, Huang P et al. G alpha(o) is necessary for muscarinic regulation of Ca2+ channels in mouse heart. Proc Natl Acad Sci USA 1997; 94: 1727–1732.
Wang Y, Park S, Bajpayee NS, Nagaoka Y, Boulay G, Birnbaumer L et al. Augmented glucose-induced insulin release in mice lacking G(o2), but not G(o1) or G(i) proteins. Proc Natl Acad Sci USA 2011; 108: 1693–1698.
Ho MK, Wong YH . G(z) signaling: emerging divergence from G(i) signaling. Oncogene 2001; 20: 1615–1625.
Ayoub MA, Damian M, Gespach C, Ferrandis E, Lavergne O, De Wever O et al. Inhibition of heterotrimeric G protein signaling by a small molecule acting on Galpha subunit. J Biol Chem 2009; 284: 29136–29145.
Holler C, Freissmuth M, Nanoff C . G proteins as drug targets. Cell Mol Life Sci 1999; 55: 257–270.
Kimple AJ, Bosch DE, Giguere PM, Siderovski DP . Regulators of G-protein signaling and their Galpha substrates: promises and challenges in their use as drug discovery targets. Pharmacol Rev 2011; 63: 728–749.
Marti-Solano M, Guixa-Gonzalez R, Sanz F, Pastor M, Selent J . Novel insights into biased agonism at G protein-coupled receptors and their potential for drug design. Curr Pharm Des 2013; 19: 5156–5166.
Donath MY, Schumann DM, Faulenbach M, Ellingsgaard H, Perren A, Ehses JA . Islet inflammation in type 2 diabetes: from metabolic stress to therapy. Diabetes Care 2008; 31 (Suppl 2): S161–S164.
Gordillo-Moscoso A, Ruiz E, Carnero M, Reguillo F, Rodriguez E, Tejerina T et al. Relationship between serum levels of triglycerides and vascular inflammation, measured as COX-2, in arteries from diabetic patients: a translational study. Lipids Health Dis 2013; 12: 62.
Vaarala O, Yki-Jarvinen H . Diabetes: should we treat infection or inflammation to prevent T2DM? Nat Rev Endocrinol 2012; 8: 323–325.
Hwang D . Fatty acids and immune responses–a new perspective in searching for clues to mechanism. Annu Rev Nutr 2000; 20: 431–456.
Luo P, Wang MH . Eicosanoids, beta-cell function, and diabetes. Prostaglandins Other Lipid Mediat 2011; 95: 1–10.
Metz SA, McRae JR, Robertson RP . Hypothesis: prostaglandins mediate defective glucose recognition in diabetes mellitus. Prostaglandins Med 1980; 4: 247–254.
Robertson RP, Chen M . A role for prostaglandin E in defective insulin secretion and carbohydrate intolerance in diabetes mellitus. J Clin Invest 1977; 60: 747–753.
Robertson RP . Eicosanoids as pluripotential modulators of pancreatic islet function. Diabetes 1988; 37: 367–370.
American Diabetes Association Task Force for Writing Nutrition P, Recommendations for the Management of D, Related C. American Diabetes Association position statement: evidence-based nutrition principles and recommendations for the treatment and prevention of diabetes and related complications. J Am Diet Assoc 2002; 102: 109–118.
Simopoulos AP . The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp Biol Med (Maywood) 2008; 233: 674–688.
Wu JH, Micha R, Imamura F, Pan A, Biggs ML, Ajaz O et al. Omega-3 fatty acids and incident type 2 diabetes: a systematic review and meta-analysis. Br J Nutr 2012; 107 (Suppl 2): S214–S227.
Bellenger J, Bellenger S, Bataille A, Massey KA, Nicolaou A, Rialland M et al. High pancreatic n-3 fatty acids prevent STZ-induced diabetes in fat-1 mice: inflammatory pathway inhibition. Diabetes 2011; 60: 1090–1099.
Wei D, Li J, Shen M, Jia W, Chen N, Chen T et al. Cellular production of n-3 PUFAs and reduction of n-6-to-n-3 ratios in the pancreatic beta-cells and islets enhance insulin secretion and confer protection against cytokine-induced cell death. Diabetes 2010; 59: 471–478.
Sorli CH, Zhang HJ, Armstrong MB, Rajotte RV, Maclouf J, Robertson RP . Basal expression of cyclooxygenase-2 and nuclear factor-interleukin 6 are dominant and coordinately regulated by interleukin 1 in the pancreatic islet. Proc Natl Acad Sci USA 1998; 95: 1788–1793.
Fujita H, Kakei M, Fujishima H, Morii T, Yamada Y, Qi Z et al. Effect of selective cyclooxygenase-2 (COX-2) inhibitor treatment on glucose-stimulated insulin secretion in C57BL/6 mice. Biochem Biophys Res Commun 2007; 363: 37–43.
Persaud SJ, Burns CJ, Belin VD, Jones PM . Glucose-induced regulation of COX-2 expression in human islets of Langerhans. Diabetes 2004; 53 (Suppl 1): S190–S192.
Shanmugam N, Todorov IT, Nair I, Omori K, Reddy MA, Natarajan R . Increased expression of cyclooxygenase-2 in human pancreatic islets treated with high glucose or ligands of the advanced glycation endproduct-specific receptor (AGER), and in islets from diabetic mice. Diabetologia 2006; 49: 100–107.
Yang F, Bleich D . Transcriptional regulation of cyclooxygenase-2 gene in pancreatic beta-cells. J Biol Chem 2004; 279: 35403–35411.
Becker RC . COX-2 inhibitors. Tex Heart Inst J 2005; 32: 380–383.
Smith CJ, McKay GA, Fisher M . Diabetes, colorectal cancer and cyclooxygenase 2 inhibition. Int J Clin Pract 2008; 62: 810–815.
Gilgore SG, Rupp JJ . The long-term response of diabetes mellitus to salicylate therapy: report of a case. JAMA 1962; 180: 65–66.
Goldfine AB, Fonseca V, Jablonski KA, Chen YD, Tipton L, Staten MA et al. Salicylate (salsalate) in patients with type 2 diabetes: a randomized trial. Ann Intern Med 2013; 159: 1–12.
Zhang J, Zou F, Tang J, Zhang Q, Gong Y, Wang Q et al. Cyclooxygenase-2-derived prostaglandin E2 promotes injury-induced vascular neointimal hyperplasia through the E-prostanoid 3 receptor. Circ Res 2013; 113: 104–114.
Chen L, Miao Y, Zhang Y, Dou D, Liu L, Tian X et al. Inactivation of the E-prostanoid 3 receptor attenuates the angiotensin II pressor response via decreasing arterial contractility. Arterioscler Thromb Vasc Biol 2012; 32: 3024–3032.
Sanchez-Alavez M, Klein I, Brownell SE, Tabarean IV, Davis CN, Conti B et al. Night eating and obesity in the EP3R-deficient mouse. Proc Natl Acad Sci USA 2007; 104: 3009–3014.
Ram A, Pandey HP, Matsumura H, Kasahara-Orita K, Nakajima T, Takahata R et al. CSF levels of prostaglandins, especially the level of prostaglandin D2, are correlated with increasing propensity towards sleep in rats. Brain Res 1997; 751: 81–89.
Dumont I, Peri KG, Hardy P, Hou X, Martinez-Bermudez AK, Molotchnikoff S et al. PGE2, via EP3 receptors, regulates brain nitric oxide synthase in the perinatal period. Am J Physiol 1998; 275: R1812–R1821.
Tilly P, Charles AL, Ludwig S, Slimani F, Gross S, Meilhac O et al. Blocking the EP3 receptor for PGE2 with DG-041 decreases thrombosis without impairing haemostatic competence. Cardiovasc Res 2014; 101: 482–491.
Robertson RP, Beiter HL, Porte D Jr . Abnormal alpha-adrenergic mechanisms in diabetes mellitus. Lancet 1973; 1: 370.
Rosengren AH, Braun M, Mahdi T, Andersson SA, Travers ME, Shigeto M et al. Reduced insulin exocytosis in human pancreatic beta-cells with gene variants linked to type 2 diabetes. Diabetes 2012; 61: 1726–1733.
Talmud PJ, Cooper JA, Gaunt T, Holmes MV, Shah S, Palmen J et al. Variants of ADRA2A are associated with fasting glucose, blood pressure, body mass index and type 2 diabetes risk: meta-analysis of four prospective studies. Diabetologia 2011; 54: 1710–1719.
Kurnik D, Muszkat M, Li C, Sofowora GG, Friedman EA, Scheinin M et al. Genetic variations in the alpha(2A)-adrenoreceptor are associated with blood pressure response to the agonist dexmedetomidine. Circ Cardiovasc Genet 2011; 4: 179–187.
Henry RR . Type 2 diabetes care: the role of insulin-sensitizing agents and practical implications for cardiovascular disease prevention. Am J Med 1998; 105: 20S–26SS.
Tayel SI, Khader HF, El-Helbawy NG, Ibrahim WA . Association of deletion allele of insertion/deletion polymorphism in alpha2B adrenoceptor gene and hypertension with or without type 2 diabetes mellitus. Appl Clin Genet 2012; 5: 111–118.
Mulder H, Nagorny CL, Lyssenko V, Groop L . Melatonin receptors in pancreatic islets: good morning to a novel type 2 diabetes gene. Diabetologia 2009; 52: 1240–1249.
Peschke E . Melatonin, endocrine pancreas and diabetes. J Pineal Res 2008; 44: 26–40.
Faria JA, Kinote A, Ignacio-Souza LM, de Araujo TM, Razolli DS, Doneda DL et al. Melatonin acts through MT1/MT2 receptors to activate hypothalamic Akt and suppress hepatic gluconeogenesis in rats. Am J Physiol Endocrinol Metab 2013; 305: E230–E242.
Zanuto R, Siqueira-Filho MA, Caperuto LC, Bacurau RF, Hirata E, Peliciari-Garcia RA et al. Melatonin improves insulin sensitivity independently of weight loss in old obese rats. J Pineal Res 2013; 55: 156–165.
McMullan CJ, Schernhammer ES, Rimm EB, Hu FB, Forman JP . Melatonin secretion and the incidence of type 2 diabetes. JAMA 2013; 309: 1388–1396.
Bouatia-Naji N, Bonnefond A, Cavalcanti-Proenca C, Sparso T, Holmkvist J, Marchand M et al. A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat Genet 2009; 41: 89–94.
Prokopenko I, Langenberg C, Florez JC, Saxena R, Soranzo N, Thorleifsson G et al. Variants in MTNR1B influence fasting glucose levels. Nat Genet 2009; 41: 77–81.
Huopio H, Cederberg H, Vangipurapu J, Hakkarainen H, Paakkonen M, Kuulasmaa T et al. Association of risk variants for type 2 diabetes and hyperglycemia with gestational diabetes. Eur J Endocrinol 2013; 169: 291–297.
Zhang C, Bao W, Rong Y, Yang H, Bowers K, Yeung E et al. Genetic variants and the risk of gestational diabetes mellitus: a systematic review. Hum Reprod Update 2013; 19: 376–390.
Bilkei-Gorzo A . The endocannabinoid system in normal and pathological brain ageing. Philos Trans R Soc Lond B Biol Sci 2012; 367: 3326–3341.
Scheen AJ, Finer N, Hollander P, Jensen MD, Van Gaal LF, Group RI-DS. Efficacy and tolerability of rimonabant in overweight or obese patients with type 2 diabetes: a randomised controlled study. Lancet 2006; 368: 1660–1672.
Pacher P, Kunos G . Modulating the endocannabinoid system in human health and disease—successes and failures. FEBS J 2013; 280: 1918–1943.
LoVerme J, Duranti A, Tontini A, Spadoni G, Mor M, Rivara S et al. Synthesis and characterization of a peripherally restricted CB1 cannabinoid antagonist, URB447, that reduces feeding and body-weight gain in mice. Bioorg Med Chem Lett 2009; 19: 639–643.
Cluny NL, Vemuri VK, Chambers AP, Limebeer CL, Bedard H, Wood JT et al. A novel peripherally restricted cannabinoid receptor antagonist, AM6545, reduces food intake and body weight, but does not cause malaise, in rodents. Br J Pharmacol 2010; 161: 629–642.
Tam J, Vemuri VK, Liu J, Batkai S, Mukhopadhyay B, Godlewski G et al. Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J Clin Invest 2010; 120: 2953–2966.
Acknowledgements
MEK has received or receives grant funding or other support from the National Institutes of Health (NIH grants DK067799 and DK080845), Juvenile Diabetes Research Foundation (JDRF grant 17-2011-608), American Diabetes Association (ADA grant 1-14-BS-115) and Pharmaceutical Research and Manufacturers of America Foundation (PhRMA Foundation Translational Medicine Starter Grant). JCN is supported by a training grant from the NIH (NIH grant AG000213). AKL is supported by funding through the US Veterans Administration. PJC has received funding from the NIH (NIH grant DK076488).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Kimple, M., Neuman, J., Linnemann, A. et al. Inhibitory G proteins and their receptors: emerging therapeutic targets for obesity and diabetes. Exp Mol Med 46, e102 (2014). https://doi.org/10.1038/emm.2014.40
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2014.40
Keywords
This article is cited by
-
Study of genetic correlation between children’s sleep and obesity
Journal of Human Genetics (2020)
-
Regulation of pancreatic β-cell function and mass dynamics by prostaglandin signaling
Journal of Cell Communication and Signaling (2017)
-
Loss of Free Fatty Acid Receptor 2 leads to impaired islet mass and beta cell survival
Scientific Reports (2016)
-
Diminished mTOR signaling: a common mode of action for endocrine longevity factors
SpringerPlus (2014)
