
Abstract
The Wnt/β-catenin pathway has a role in osteoblast differentiation and bone formation. We screened 100 plant extracts and identified an extract from Euodia sutchuenensis Dode (ESD) leaf and young branch as an effective activator of the Wnt/β-catenin pathway. ESD extract increased β-catenin levels and β-catenin nuclear accumulation in murine primary osteoblasts. The ESD extract also increased mRNA levels of osteoblast markers, including RUNX2, BMP2 and COL1A1, and enhanced alkaline phosphatase (ALP) activity in murine primary osteoblasts. Both ESD extract-induced β-catenin increment and ALP activation were abolished by β-catenin knockdown, confirming that the Wnt/β-catenin pathway functions in osteoblast differentiation. ESD extract enhanced terminal osteoblast differentiation as shown by staining with Alizarin Red S and significantly increased murine calvarial bone thickness. This study shows that ESD extract stimulates osteoblast differentiation via the Wnt/β-catenin pathway and enhances murine calvarial bone formation ex vivo.
Similar content being viewed by others


Introduction
Given the rapid growth of global elderly populations, osteoporosis is becoming a major public health issue.1 Low bone mineral density and structural deterioration of bone tissue in osteoporosis leads to disability and early mortality.2, 3 Current therapeutic strategies for osteoporosis treatment use anti-resorptive agents such as estrogen and bisphosphonates to inhibit osteoclast activity.4, 5 Although estrogen replacement therapy improves bone mineral density and reduces fracture incidence in early menopausal women, long-term estrogen use increases the risks of breast cancer, abnormal uterine bleeding and cardiovascular diseases.6 Bisphosphonates, another class of anti-resorptive drug, cause severe and incapacitating bone, joint and/or muscle pains;7, 8 long-term treatment leads to severe bone fractures due to the accumulation of microfractures.9
To address the limitations of anti-resorptive drugs that inhibit osteoclast activity, anabolic agents are considered as beneficial therapeutics for osteoporosis treatment.10 Anabolic agents increase osteoblast differentiation and proliferation,11, 12 which enhances bone mass, quality, strength and composition.13, 14 Parathyroid hormone drugs such as full-length and N-terminal (1–34) fragment of parathyroid hormone are bone anabolic agents approved by the US Food and Drug Administration or the European Union.15 However, parathyroid hormone drugs have adverse side effects that potentially increase osteosarcoma and hyperparathyroidism incidence.11, 16 Parathyroid hormone drugs are relatively expensive and should be taken as daily injections.17 There is a great need to discover novel anabolic agents for osteoporosis treatment.
The Wnt/β-catenin pathway increases osteoblast differentiation and subsequent bone formation.18, 19 Nuclear accumulation of stabilized β-catenin is an important indicator of osteoblast differentiation.20, 21 Inactivation of Axin2, a negative regulator of the Wnt/β-catenin pathway, promotes osteoblast proliferation and differentiation and matrix mineralization in vitro and in vivo.22 Activation of the Wnt/β-catenin pathway enhances Runx2 expression, which is a crucial transcription factor for osteoblast differentiation.23, 24 Therefore, discovery and development of drugs that activate the Wnt/β-catenin pathway is a promising approach for the development of novel osteoporosis therapies. Glycogen synthase kinase-3β inhibitors or antibodies against Wnt/β-catenin pathway antagonists, including sclerostin, secreted frizzled-related protein-1 and dickkopf-1, induce bone formation in mice, rats or humans.25, 26 None of these drugs are currently approved for clinical applications. Natural products, including plant extracts, are traditionally used to treat diseases and are often used for drug development owing to fewer side effects compared with those of synthetic compounds.27 Plants such as Eurycoma longifolia and Labisia pumila are used as traditional East Asian medicines for bone diseases, including osteoporosis.28
Here we screened 100 plant extracts for the ability to activate the Wnt/β-catenin pathway in HEK293 cells harboring a chromosomal Wnt/β-catenin signaling reporter and characterized the effects on osteoblast differentiation and murine calvarial bone formation. We identified an extract from the leaf and young branch of Euodia sutchuenensis Dode (ESD) as an activator of the Wnt/β-catenin pathway. Activation of the Wnt/β-catenin pathway by ESD extract was confirmed, and its effect on osteoblast differentiation was demonstrated using murine primary osteoblasts. The effect of ESD extract in bone formation was investigated by ex vivo measuring murine calvarial bone formation. These results show that ESD extract is a potential Wnt/β-catenin pathway agent that can be used for osteoporosis treatment.
Materials and methods
Materials
A library containing methanol extracts of 100 plants was purchased from International Biological Material Research Center, Korea Research Institute of Bioscience and Biotechnology, Daejeon, Korea. The library contained extracts from Vietnamese plants, including an extract from the leaf and young branch of ESD. Extracts were dissolved in dimethyl sulfoxide (DMSO; Sigma Aldrich, St Louis, MO, USA).
Cell culture
HEK293-reporter cells containing chromosomally integrated TCF reporter (TOPflash)29 were grown in Dulbecco’s Modified Eagle Medium (Gibco BRL, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Gibco BRL) and 100 U ml−1 penicillin/streptomycin (Gibco BRL). Murine primary osteoblasts were obtained from 4-day-old imprinting control region mice. Calvaria were dissected and digested five times in sequence with 0.32 mg ml−1 collagenase type II (Worthington, Lakewood, NJ, USA) for 20 min at 37 °C. Cells were cultured in α-minimal essential medium (Gibco BRL) supplemented with 10% fetal bovine serum and 100 U ml−1 penicillin/streptomycin (Gibco BRL). For osteoblast cell differentiation, 50 μg ml−1 ascorbic acid (Sigma Aldrich) and 10 mM β-glycerophosphate (Sigma Aldrich) were added to the α-minimal essential medium.
Luciferase reporter assay
HEK293-reporter cells were seeded at a density of 2.7 × 104 cells per well in 96-well white polystyrene plates (Greiner Bio-One, Stonehouse, UK). Cells were treated with each plant extract, DMSO (negative control) or Hovenia dulcis Thunb. (HDT, positive control30) at a final concentration of 5 μg ml−1 for 24 h. TOPflash reporter activity was measured as described previously,29 and relative reporter activity was calculated by normalizing to the DMSO negative control. For quantitative analysis of TOPflash reporter activity induced by ESD extract, HEK293-reporter cells were seeded in 24-well plate at a density of 7 × 104 cells per well, and cells were treated with ESD extract (1 or 5 μg ml−1) for 24 h. Total cell lysates were extracted with 55 μl of 5 × Reporter Lysis Buffer (Promega, Madison, WI, USA) per well. Next, 15 μl of luciferin was added to 30 μl of cell lysate, and luciferase activity was measured using the FLUOstar Optima plate reader (BMG Labtech, Offenburg, Germany).
Cell viability assay
Murine primary osteoblasts were seeded in 24-well plates at a density of 2.5 × 104 cells per well and treated with DMSO or ESD extract for 72 h. Next, 0.25 mg ml−1 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Amresco, Solon, OH, USA) was added to each well, cells were incubated for 2 h at 37 °C, and the insoluble formazan product was obtained by removal of the medium. The formazan product was dissolved by adding 1 ml DMSO for 30 min and then quantitated by reading the absorbance at 590 nm.30
Immunoblotting
Murine primary osteoblasts were rinsed with ice-cold phosphate-buffered saline (PBS; Gibco BRL), lysed in 70 μl of radioimmunoprecipitation assay buffer (Millipore, Bedford, MA, USA), incubated for 10 min at 4 °C and centrifuged at 15 920 g for 30 min. Proteins were separated to 8% sodium dodecyl sulphate–polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Whatman, Florham Park, NJ, USA). Next, proteins were incubated with the following primary antibodies: anti-β-catenin (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and anti-α-tubulin (Oncogene Research Products, Cambridge, MA, USA). Secondary antibody was horseradish peroxidase-conjugated anti-mouse (Cell Signaling, Beverly, MA, USA) or anti-rabbit (Bio-Rad Laboratories, Hercules, CA, USA). Proteins were visualized using enhanced chemiluminescence (Amersham Bioscience, Piscataway, NJ, USA) and a luminescent image analyzer (LAS-3000; Fujifilm, Tokyo, Japan).
Immunocytochemistry
Murine primary osteoblasts were seeded on glass coverslips in a 12-well plate at a density of 4 × 104 cells per well and treated with DMSO or ESD extract for 24 h at 37 °C. Next, cells were fixed with 4% paraformaldehyde for 15 min and permeabilized with 0.1% Triton X-100 for 15 min. Cells were blocked with 5% bovine serum albumin for 1 h and incubated with anti-β-catenin antibody (Santa Cruz Biotechnology) overnight at 4 °C. Cells were then rinsed with PBS, incubated with anti-mouse Alexa Flour 488 (Life Technologies, Carlsbad, CA, USA) secondary antibody for 1 h and counterstained with 4′,6-diamidino-2-phenylindole (Sigma Aldrich). Cell fluorescence was observed and captured using a confocal microscope (LSM510; Carl Zeiss Inc., Thornwood, NY, USA). Measurement of β-catenin fluorescence intensity was performed using the NIS elements AR 3.1 software (Nikon, Tokyo, Japan).
RNA extraction and quantitative real-time PCR
For RNA extraction, murine primary osteoblasts were seeded in six-well plates at a density of 9 × 104 cells per well and cultured for 2 days. Next, ESD extract in differentiation medium was added, and cells were incubated for 48 h as described in the Cell culture section. Total RNA was prepared using TRIzol (Invitrogen, Carlsbad, CA, USA), and 4 μg of total RNA was reverse transcribed into cDNA using 200 U of reverse transcriptase (Invitrogen) according to the manufacturer’s instructions. The gene expression was normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression as a house keeping gene. The following primers were used: bone morphogenetic protein 2 (BMP2), forward 5′-ATCCAGTCTTGCCGCCTC-3′ and reverse 5′-GCCTCCTCCTCCTTCTCC-3′; runt-related transcription factor 2 (RUNX2), forward 5′-AAGGACAGAGTCAGATTACAGA-3′ and reverse 5′-GTGGTGGAGTGGATGGAT-3′; collagen type I alpha 1 (COL1A1), forward 5′-GGTCTTCCAGGTCCTAAGG-3′ and reverse 5′-CACGGGCACCATCTTTAC-3′; and GAPDH, forward 5′-CCATGGAGAAGGCTGGGG-3′ and reverse 5′-CAAAGTTGTCATGGATGACC-3′.
Alkaline phosphatase (ALP) staining
Murine primary osteoblasts were seeded at a density of 5 × 104 cells per well in a 24-well plate and cultured for 2 days. Next, ESD extract in differentiation medium was added, and cells were incubated for 48 h as described in the Cell culture section. For the ALP staining assay, ALP activites were visualized using the TRACP and ALP Double-Stain kit (Takara Bio Inc., Shiga, Japan) according to the manufacturer’s instructions. Images were visualized with a bright field optical microscope (Eclipse TE2000-U; Nikon).
ALP assay
Murine primary osteoblasts were seeded in a 24-well plate at a density of 5 × 104 cells per well and cultured for 2 days. Next, ESD extract in differentiation medium was added, and cells were incubated for 48 h as described in the Cell culture section. To measure ALP activity, cells were rinsed with PBS and lysed with 55 μl of 5 × Reporter Lysis Buffer (Promega) per well. Cells were incubated for 10 min at 4 °C and centrifuged at 15 920 g for 30 min at 4 °C. The supernatant (20 μl) was incubated with 100 μl Tris-glycine buffer pH 10.3 (50 mM Tris-HCl (Amresco), 100 mM glycine (Duksan, Ansan, Gyeonggido, Korea) and 2 mM MgCl2 (Duksan)) and 100 μl of p-nitrophenyl phosphate (Sigma Aldrich). The reaction was stopped with 50 μl of 3 M NaOH, and the absorbance was measured at 405 nm.31 ALP activity was quantified by measuring the protein concentration using the Bradford assay (Bio-Rad Laboratories).
Alizarin Red S staining
Murine primary osteoblasts were seeded in a 24-well plate at a density of 5 × 104 cells per well and cultured for 2 days. Next, ESD extract in differentiation medium was added, and cells were incubated for 14 days as described in the Cell culture section. Medium was changed every 2 days. After 14 days, cells were rinsed with cold PBS, fixed with 4% paraformaldehyde for 20 min at room temperature and stained with 2% Alizarin red S staining solution (pH 4.2; Sigma Aldrich) for 30 min at room temperature. For quantification, Alizarin Red was solubilized with 10% acetic acid, followed by heating at 85 °C for 10 min. The eluates were neutralized in 10% ammonium hydroxide. The quantity of Alizarin Red was measured spectrophotometrically at 405 nm.26 Images were recorded with a bright field optical microscope (Eclipse TE2000-U; Nikon).
Calvaria ex vivo culture and hematoxylin and eosin staining
Calvaria isolated from 4-day-old imprinting control region mice were grown in a 12-well plate with α-minimal essential medium for 24 h and treated with ESD extract in differentiation medium for 7 days. Medium was changed every 2 days. Calvaria were fixed in 4% paraformaldehyde for 2 days and then decalcified in 4% formic acid and 4% hydrochloric acid for 2 days at 4 °C. Next, calvaria were embedded in paraffin, sectioned in 4-μm thickness (Leica Microsystems, Wetzlar, Germany). The 4-μm bone tissue sections were deparaffinized in xylene and in a graded ethanol series and used for hematoxylin and eosin orange G staining. The hematoxylin and eosin-stained preparations were visualized with a bright field optical microscope (Eclipse TE2000-U; Nikon).
Immunohistochemistry
For florescence immunohistochemistry analyses, the 4-μm bone tissue sections were deparaffinized in xylene and in a graded ethanol series and incubated for 4 h at 60 °C in sodium citrate buffer (10 mM sodium citrate, pH 6.0) for antigen retrieval. The sections were blocked in 5% bovine serum albumin and 1% goat serum in PBS at room temperature for 1 h and then incubated with primary antibody anti-β-catenin (Santa Cruz Biotechnology) and secondary antibody anti-mouse Alexa Flour 555 (Life Technologies). The sections were stained with 4′,6-diamidino-2-phenylindole and mounted in Gel/Mount medium (Biomeda Corporation, Foster City, CA, USA). A confocal microscopy (LSM510; Carl Zeiss Inc.) observed and captured fluorescence.30
β-Catenin knockdown mediated by small interfering RNA (siRNA) transfection
Murine primary osteoblasts were seeded in six-well plates at a density of 1 × 105 cells per well. The cells were transfected with siRNA for β-catenin or negative control (Bioneer, Daejeon, Korea) using Lipofectamine Plus (Invitrogen) in serum-free Opti-MEM (Gibco BRL) according to the manufacturer’s instructions. After 12-h transfection with β-catenin siRNA, ESD extract in differentiation medium was added, and cells were incubated for 48 h before harvesting. ALP assay and immunoblotting were performed to examine changes in ALP activity and to confirm the transfection efficiency of β-catenin siRNA.
Statistical analyses
Each experiment was performed at least three times, and results were presented as means±s.d. Statistical significance was analyzed using the Student’s t-test for experiments involving two groups. P-values <0.05 were considered statistically significant (*P<0.05, **P<0.01, ***P<0.001).
Results
Identification of ESD extract as a Wnt/β-catenin pathway activator
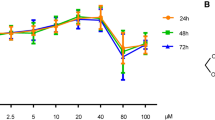
We screened a library containing methanol extracts of 100 plants using HEK293-reporter cells to identify plant extracts that stimulate the Wnt/β-catenin pathway (Figure 1a). ESD and Impatiens eberhardtii Tardieu extracts produced >140% TOPflash reporter activity compared with that of control (Figure 1a). We selected ESD extract for further analysis due to 15% greater increase in TOPflash activity compared with that of Impatiens eberhardtii Tardieu or HDT (positive control) (Figure 1b).30 Furthermore, ESD extract increased TOPflash activity in a dose-dependent manner (Figure 1c).

Screening of a library of 100 plant extracts for Wnt/β-catenin pathway activators. HEK293-reporter cells were treated with extracts for 24 h, and cell lysates were subjected to TOPflash reporter assay (n=3). (a) A library of 100 plant extracts was screened. (b) HEK293-reporter cells were treated with extracts from Euodia sutchuenensis Dode (ESD) or Impatiens eberhardtii Tardieu (IET), which produced >140% TOPflash activity compared with controls; Hovenia dulcis Thunb. (HDT) extract was used as a positive control. (c) HEK293-reporter cells were treated with the ESD extract at different concentrations, and activation of the Wnt/β-catenin pathway was measured. **P<0.01, ***P<0.001 versus control.
ESD extract activates the Wnt/β-catenin pathway in murine primary osteoblasts
ESD extract is not significantly toxic to murine primary osteoblasts (Figure 2a). To investigate whether ESD extract activates the Wnt/β-catenin pathway in murine primary osteoblasts, we examined β-catenin levels in murine primary osteoblasts treated with the ESD extract. ESD extract increased β-catenin levels in murine primary osteoblasts in a dose-dependent manner as determined by immunoblotting and immunocytochemistry (Figures 2b–d). Nuclear accumulation of β-catenin was elevated in murine primary osteoblasts treated with the ESD extract (Figures 2c and e). Together, these results indicate that ESD extract induces Wnt/β-catenin pathway activation in murine primary osteoblasts.

Effects of ESD extract on activation of the Wnt/β-catenin pathway in murine primary osteoblasts. (a) Murine primary osteoblasts were treated with various ESD extract concentrations for 72 h, and cell viability was assessed by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay (n=3). (b–e) Murine primary osteoblasts were treated with 0, 1 or 5 μg ml−1 of ESD extract for 24 h. (b) Immunoblotting analysis was performed to detect β-catenin and α-tubulin. (c) Immunofluorescence staining was performed to detect β-catenin (green). Nuclei were counterstained with DAPI (4′,6-diamidino-2-phenylindole; blue). White arrows indicate nuclear localization of β-catenin. Scale bars=100 μm. (d) Quantitative analyses of total β-catenin intensity. (e) Quantitative analysis of β-catenin nuclear accumulation (n>3). **P<0.01, ***P<0.001 versus control.
ESD extract stimulates murine primary osteoblast differentiation via Wnt/β-catenin pathway activation
As activation of the Wnt/β-catenin pathway induces osteoblast differentiation, we examined the effect of ESD extract on murine primary osteoblast differentiation. Transcriptional levels of osteogenic differentiation marker genes such as BMP2, RUNX2 and COL1A1 were approximately 1.5- to 2-fold higher after treatment with ESD extract compared with those of controls (Figure 3a). ESD extract also enhanced ALP activity as determined by ALP staining and direct measurement of ALP activity (Figures 3b and c). Furthermore, ESD extract enhanced terminal osteoblast differentiation as determined by Alizarin Red S staining in murine primary osteoblasts (Figures 4a and b).

Effects of ESD extract on osteogenic differentiation of murine primary osteoblasts. (a–c) Murine primary osteoblasts were treated with 5 μg ml−1 ESD extract or DMSO (control) in differentiation medium for 48 h. (a) Real-time reverse transcriptase-PCR assay was performed to examine changes in osteoblast marker gene expression, RUNX2, BMP2 and COL1A1. Relative gene expression was normalized to GAPDH expression (n=3). (b) Cells were subjected to alkaline phosphatase (ALP) staining. (c) Cells were subjected to ALP activity measurement; ALP activity was normalized with respect to total protein concentration. *P<0.05, **P<0.01 versus control.

Effects of ESD extract on terminal osteogenic differentiation of murine primary osteoblasts. (a, b) Murine primary osteoblasts were treated with ESD extract for 14 days. (a) Matrix mineralization was visualized by Alizarin Red S staining. (b) Alizarin Red S staining was quantified by measuring absorbance at 450 nm. *P<0.05 versus control.
To verify Wnt/β-catenin pathway activation in ESD extract-induced osteogenic differentiation, we measured the effects of ESD extract on osteogenic differentiation of murine primary osteoblasts when β-catenin was knocked down. ESD extract-enhanced ALP activity was abolished by β-catenin knockdown (Figures 5a and b). Taken together, these results indicate that ESD extract activates osteoblast differentiation by upregulating the Wnt/β-catenin pathway.

Effects of ESD extract on osteogenic differentiation of murine primary osteoblasts via Wnt/β-catenin pathway activation. (a–b) Murine primary osteoblasts were transfected with β-catenin siRNA or control siRNA for 12 h and then treated with 5 μg ml−1 ESD extract in differentiation media for 48 h. (a) Immunoblotting analyses were performed to detect β-catenin and α-tubulin. (b) Cells were subjected to ALP activity measurement; ALP activity was normalized with respect to the total protein concentration (n>3). **P<0.01 versus control.
ESD extract increases murine calvarial thickness ex vivo
To investigate whether ESD extract-induced osteogenic differentiation in vitro promoted bone formation ex vivo, calvaria isolated from 4-day-old mice were treated with ESD extract for 7 days. Hematoxylin and eosin staining showed that ESD extract significantly increased the thickness of murine calvarial bone in an ex vivo culture system (Figures 6a and b). ESD extract increased the number of β-catenin-positive cells (Figures 6c and d) in murine calvaria. These results indicate that ESD extract enhances murine calvarial bone formation ex vivo together with activation of the Wnt/β-catenin pathway.

Effects of ESD extract on murine calvaria bone formation ex vivo. (a–d) Murine calvaria were treated with 5 μg ml−1 ESD extract in osteogenic differentiation medium for 7 days. (a) Hematoxylin and eosin staining was performed to evaluate calvaria (original magnification × 400). (b) Bone thickness was quantified (n=3). (c) Calvaria were fluorescently stained for β-catenin. White arrows indicate β-catenin-positive cells. Scale bars=100 μm. (d) β-Catenin-positive cells were quantified as the number of β-catenin-positive cells divided by the total number of cells (n>3). *P<0.05, **P<0.01 versus control.
Discussion
Bone remodeling is a dynamic and continuous process that maintains a constant bone mass.32 It is regulated by a balance between osteoblast-mediated formation of new bone and osteoclast-mediated removal of old bone.3, 33 An imbalance in bone formation and resorption causes bone-related diseases, including osteoporosis. Bone density tends to decrease during aging,34 leading to weaker bone, elevated bone loss and greater risk of catastrophic bone fracture. Postmenopausal estrogen deficiency frequently leads to accelerated bone loss and osteoporosis in women.35 Age-related and postmenopausal osteoporosis is commonly associated with osteoblast dysfunction.36, 37 Conversely, stimulating osteoblast function promotes bone formation and increases bone strength.38 New therapeutic approaches for osteoporosis require identification and development of new anabolic agents that improve bone mass by stimulating osteoblast activity.39
Several signaling pathways regulate osteoblast differentiation, including Wnt,18, 19 parathyroid hormone40 and bone morphogenic protein.41 The role of Wnt/β-catenin signaling in osteoblast differentiation and bone formation has attracted considerable interest in recent years.25 Aberrant activation of the Wnt/β-catenin pathway is implicated in tumorigenesis;42 however, a recent report shows that this pathway is inactive in osteosarcoma cell lines.43 Activation of the Wnt/β-catenin pathway stimulates osteogenic differentiation and inhibits cell proliferation in osteosarcoma cell lines.43 Therefore, identification of drugs that directly activate the Wnt/β-catenin pathway is a promising approach to identify drugs for anabolic bone formation.
We screened a library of plant extracts and identified an extract from the leaf and young branch of ESD as an effective activator of the Wnt/β-catenin pathway. ESD is an ornamental tree that belongs to Rutaceae, occurring in temperate regions. In Korea and China, ESD has been used as a herbal medicine to relieve headache, gastric inflammation and dermatitis.44 ESD has also been reported to have anti-feedant activity mediated by the linear furanocoumarins and anti-mycobacterial activity mediated by the geranylated furanocoumarins isolated from its fruits.45, 46 However, the effect of ESD on osteoblast differentiation has not been revealed. ESD extract-induced osteoblast differentiation was confirmed by ALP activation and induction of RUNX2, BMP2 and COL1A1 expression. The effect of ESD extract on bone formation was further confirmed by enhanced mineralization and calvarial bone formation. The function of ESD extract in bone formation was verified ex vivo by measuring the number of β-catenin-positive cells in murine calvaria (Figures 5c and d). The leaf of ESD contains ingredients such as evodioside B, hesperidin, limonin, simplexoside, vitexin, uracil and myo-inositol.47 Hesperidin is a monomethoxylated flavanone that exists in citrus fruits, and several studies have shown that hesperidin inhibits bone loss.48, 49 Myo-inositol has been reported to have essential roles in osteogenesis and bone formation.50 Therefore, it is possible that hesperidin and myo-inositol in ESD may have roles in the enhancement of osteogenic differentiation.
Taken together, our data show that ESD extract stimulates osteoblast differentiation in vitro, increases the thickness of mice calvarial bone ex vivo and activates Wnt/β-catenin signaling. These results suggest that ESD extract might have therapeutic potential for clinical treatment of osteoporosis.
References
Cheung AM, Feig DS, Kapral M, Diaz-Granados N, Dodin S . Canadian Task Force on Preventive Health Care. Prevention of osteoporosis and osteoporotic fractures in postmenopausal women: recommendation statement from the Canadian Task Force on Preventive Health Care. CMAJ 2004; 170: 1665–1667.
Old JL, Calvert M . Vertebral compression fractures in the elderly. Am Fam Physician 2004; 69: 111–116.
Khosla S, Riggs BL . Pathophysiology of age-related bone loss and osteoporosis. Endocrinol Metab Clin North Am 2005; 34: 1015–1030.
Hellstein JW, Adler RA, Edwards B, Jacobsen PL, Kalmar JR, Koka S et al. Managing the care of patients receiving antiresorptive therapy for prevention and treatment of osteoporosis: executive summary of recommendations from the American Dental Association Council on Scientific Affairs. J Am Dent Assoc 2011; 142: 1243–1251.
Lee YJ, Jeong JK, Seol JW, Xue M, Jackson C, Park SY . Activated protein C differentially regulates both viability and differentiation of osteoblasts mediated by bisphosphonates. Exp Mol Med 2013; 45: e9.
Tang DZ, Hou W, Zhou Q, Zhang M, Holz J, Sheu TJ et al. Osthole stimulates osteoblast differentiation and bone formation by activation of beta-catenin-BMP signaling. J Bone Miner Res 2010; 25: 1234–1245.
Licata AA . Discovery, clinical development, and therapeutic uses of bisphosphonates. Ann Pharmacother 2005; 39: 668–677.
Rotella DP . Osteoporosis: challenges and new opportunities for therapy. Curr Opin Drug Discov Devel 2002; 5: 477–486.
Kennel KA, Drake MT . Adverse effects of bisphosphonates: implications for osteoporosis management. Mayo Clin Proc 2009; 84: 632–637 quiz 638.
Guo AJ, Choi RC, Cheung AW, Chen VP, Xu SL, Dong TT et al. Baicalin, a flavone, induces the differentiation of cultured osteoblasts: an action via the Wnt/beta-catenin signaling pathway. J Biol Chem 2011; 286: 27882–27893.
Khan AW, Khan A . Anabolic agents: a new chapter in the management of osteoporosis. J Obstet Gynaecol Can 2006; 28: 136–141.
Tang DZ, Yang F, Yang Z, Huang J, Shi Q, Chen D et al. Psoralen stimulates osteoblast differentiation through activation of BMP signaling. Biochem Biophys Res Commun 2011; 405: 256–261.
Sosa M, González-Padilla E . Promising developments in osteoporosis treatment. Int J Clin Rheumatol 2011; 6: 325–332.
Sibai T, Morgan EF, Einhorn TA . Anabolic agents and bone quality. Clin Orthop Relat Res 2011; 469: 2215–2224.
Rachner TD, Khosla S, Hofbauer LC . Osteoporosis: now and the future. Lancet 2011; 377: 1276–1287.
Tashjian AH Jr, Gagel RF . Teriparatide [human PTH(1-34)]: 2.5 years of experience on the use and safety of the drug for the treatment of osteoporosis. J Bone Miner Res 2006; 21: 354–365.
Garrett IR . Anabolic agents and the bone morphogenetic protein pathway. Curr Top Dev Biol 2007; 78: 127–171.
Liu F, Kohlmeier S, Wang CY . Wnt signaling and skeletal development. Cell Signal 2008; 20: 999–1009.
Piters E, Boudin E, Van Hul W . Wnt signaling: a win for bone. Arch Biochem Biophys 2008; 473: 112–116.
Baron R, Rawadi G, Roman-Roman S . Wnt signaling: a key regulator of bone mass. Curr Top Dev Biol 2006; 76: 103–127.
Zhang J, Zhang X, Zhang L, Zhou F, van Dinther M, Ten Dijke P . LRP8 mediates Wnt/beta-catenin signaling and controls osteoblast differentiation. J Bone Miner Res 2012; 27: 2065–2074.
Yu HM, Jerchow B, Sheu TJ, Liu B, Costantini F, Puzas JE et al. The role of Axin2 in calvarial morphogenesis and craniosynostosis. Development 2005; 132: 1995–2005.
Gaur T, Lengner CJ, Hovhannisyan H, Bhat RA, Bodine PV, Komm BS et al. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J Biol Chem 2005; 280: 33132–33140.
Komori T . Regulation of skeletal development by the Runx family of transcription factors. J Cell Biochem 2005; 95: 445–453.
Hoeppner LH, Secreto FJ, Westendorf JJ . Wnt signaling as a therapeutic target for bone diseases. Expert Opin Ther Targets 2009; 13: 485–496.
Zahoor M, Cha PH, Min do S, Choi KY . Indirubin-3'-oxime reverses bone loss in ovariectomized and hindlimb-unloaded mice via activation of the Wnt/beta-catenin signaling. J Bone Miner Res 2014; 29: 1196–1205.
Newman DJ, Cragg GM . Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod 2012; 75: 311–335.
Abd Jalil MA, Shuid AN, Muhammad N . Role of medicinal plants and natural products on osteoporotic fracture healing. Evid Based Complement Alternat Med 2012; 2012: 714512.
Yun MS, Kim SE, Jeon SH, Lee JS, Choi KY, Both ERK . and Wnt/beta-catenin pathways are involved in Wnt3a-induced proliferation. J Cell Sci 2005; 118: 313–322.
Cha PH, Shin W, Zahoor M, Kim HY, Min do S, Choi KY . Hovenia dulcis Thunb extract and its ingredient methyl vanillate activate Wnt/beta-catenin pathway and increase bone mass in growing or ovariectomized mice. PLoS One 2014; 9: e85546.
Sila-Asna M, Bunyaratvej A, Maeda S, Kitaguchi H, Bunyaratavej N . Osteoblast differentiation and bone formation gene expression in strontium-inducing bone marrow mesenchymal stem cell. Kobe J Med Sci 2007; 53: 25–35.
Kim WK, Kim JC, Park HJ, Sul OJ, Lee MH, Kim JS et al. Platinum nanoparticles reduce ovariectomy-induced bone loss by decreasing osteoclastogenesis. Exp Mol Med 2012; 44: 432–439.
Rodan GA . Bone homeostasis. Proc Natl Acad Sci USA 1998; 95: 13361–13362.
Chung PL, Zhou S, Eslami B, Shen L, Leboff MS, Glowacki J . Effect of age on regulation of human osteoclast differentiation. J Cell Biochem 2014; 115: 1412–1419.
Miyauchi Y, Sato Y, Kobayashi T, Yoshida S, Mori T, Kanagawa H et al. HIF1alpha is required for osteoclast activation by estrogen deficiency in postmenopausal osteoporosis. Proc Natl Acad Sci USA 2013; 110: 16568–16573.
Kassem M, Marie PJ . Senescence-associated intrinsic mechanisms of osteoblast dysfunctions. Aging Cell 2011; 10: 191–197.
Albright F, Smith PH, Richardson AM . Postmenopausal osteoporosis: its clinical features. JAMA 1941; 116: 2465–2474.
Marie PJ, Kassem M . Osteoblasts in osteoporosis: past, emerging, and future anabolic targets. Eur J Endocrinol 2011; 165: 1–10.
Lane NE, Kelman A . A review of anabolic therapies for osteoporosis. Arthritis Res Ther 2003; 5: 214–222.
Isogai Y, Akatsu T, Ishizuya T, Yamaguchi A, Hori M, Takahashi N et al. Parathyroid hormone regulates osteoblast differentiation positively or negatively depending on the differentiation stages. J Bone Miner Res 1996; 11: 1384–1393.
Yamaguchi A, Ishizuya T, Kintou N, Wada Y, Katagiri T, Wozney JM et al. Effects of BMP-2, BMP-4, and BMP-6 on osteoblastic differentiation of bone marrow-derived stromal cell lines, ST2 and MC3T3-G2/PA6. Biochem Biophys Res Commun 1996; 220: 366–371.
Luu HH, Zhang R, Haydon RC, Rayburn E, Kang Q, Si W et al. Wnt/beta-catenin signaling pathway as a novel cancer drug target. Curr Cancer Drug Targets 2004; 4: 653–671.
Cai Y, Mohseny AB, Karperien M, Hogendoorn PC, Zhou G, Cleton-Jansen AM . Inactive Wnt/beta-catenin pathway in conventional high-grade osteosarcoma. J Pathol 2010; 220: 24–33.
Wiart C . Lead Compounds from Medicinal Plants for the Treatment of Neurodegenerative Diseases. Academic Press: London, UK, 2014.
Stevenson PC, Simmonds MS, Yule MA, Veitch NC, Kite GC, Irwin D et al. Insect antifeedant furanocoumarins from Tetradium daniellii. Phytochemistry 2003; 63: 41–46.
Adams M, Ettl S, Kunert O, Wube AA, Haslinger E, Bucar F et al. Antimycobacterial activity of geranylated furocoumarins from Tetradium daniellii. Planta Med 2006; 72: 1132–1135.
Yoo SW, Kim JS, Kang SS, Son KH, Chang HW, Kim HP et al. Constituents of the fruits and leaves of Euodia daniellii. Arch Pharm Res 2002; 25: 824–830.
Horcajada MN, Habauzit V, Trzeciakiewicz A, Morand C, Gil-Izquierdo A, Mardon J et al. Hesperidin inhibits ovariectomized-induced osteopenia and shows differential effects on bone mass and strength in young and adult intact rats. J Appl Physiol (1985) 2008; 104: 648–654.
Chiba H, Kim H, Matsumoto A, Akiyama S, Ishimi Y, Suzuki K et al. Hesperidin prevents androgen deficiency-induced bone loss in male mice. Phytother Res 2014; 28: 289–295.
Dai Z, Chung SK, Miao D, Lau KS, Chan AW, Kung AW . Sodium/myo-inositol cotransporter 1 and myo-inositol are essential for osteogenesis and bone formation. J Bone Miner Res 2011; 26: 582–590.
Acknowledgements
This work was supported by grants from the National Research Foundation (NRF) funded by the Ministry of Education, Science and Technology of Korea (the Mid-career Researcher Program (2012-010285), Translational Research Center for Protein Function Control; 2009-0083522) and Stem Cell Research Project (2010-0020235). This work was also supported by a grant from the Korea Research Institute of Chemical Technology (SI-095). J-H Hwang was supported by a BK21 scholarship from the Ministry of Education and Human Resources Development.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Hwang, JH., Cha, PH., Han, G. et al. Euodia sutchuenensis Dode extract stimulates osteoblast differentiation via Wnt/β-catenin pathway activation. Exp Mol Med 47, e152 (2015). https://doi.org/10.1038/emm.2014.115
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2014.115
This article is cited by
-
Mutations in ARHGEF15 cause autosomal dominant hereditary cerebral small vessel disease and osteoporotic fracture
Acta Neuropathologica (2023)
-
Tenuigenin promotes the osteogenic differentiation of bone mesenchymal stem cells in vitro and in vivo
Cell and Tissue Research (2017)
-
Bergapten promotes bone marrow stromal cell differentiation into osteoblasts in vitro and in vivo
Molecular and Cellular Biochemistry (2015)
