
Abstract
Long intergenic non-coding RNAs (lincRNAs) have historically been ignored in cancer biology. However, thousands of lincRNAs have been identified in mammals using recently developed genomic tools, including microarray and high-throughput RNA sequencing (RNA-seq). Several of the lincRNAs identified have been well characterized for their functions in carcinogenesis. Here we performed RNA-seq experiments comparing gastric cancer with normal tissues to find differentially expressed transcripts in intergenic regions. By analyzing our own RNA-seq and public microarray data, we identified 31 transcripts, including a known expressed sequence tag, BM742401. BM742401 was downregulated in cancer, and its downregulation was associated with poor survival in gastric cancer patients. Ectopic overexpression of BM742401 inhibited metastasis-related phenotypes and decreased the concentration of extracellular MMP9. These results suggest that BM742401 is a potential lincRNA marker and therapeutic target.
Similar content being viewed by others


Introduction
Long intergenic (or intervening) non-coding RNAs (lincRNAs) are encoded in genomic loci that do not overlap protein-coding genes. LincRNAs are longer than 200 nucleotides, capped, poly-adenylated and often spliced in human and mouse.1, 2 Some lincRNAs were previously characterized as non-coding RNAs (ncRNAs). Conventionally, ncRNAs have been identified by shotgun sequencing of expressed sequence tags and cloned cDNA. Microarray platforms have also been used to identify them on a genome-wide level.3, 4 Recently, using high-throughput RNA sequencing (RNA-seq) technology, researchers have identified novel transcripts not capable of being measured using conventional analyses.5, 6, 7
Using recently developed genomic tools, such as microarray and RNA-seq analysis, thousands of lincRNAs have been identified in mammals, but the functions of these lincRNAs have only been reported for a small number. Studies have revealed several important regulatory roles of lincRNAs, including X chromosome inactivation (XIST), imprinting (H19, KCNQ1OT1) and development (HOTAIR).8, 9, 10, 11 Recent studies have suggested various molecular functions of lincRNAs, including maintenance of pluripotency, p53 response pathways and transcriptional regulation by epigenetic controls.2, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21 One controversial issue in the ncRNA field is whether lincRNAs work in cis or in trans. By global screening, a few dozen lincRNAs were reported to work in trans to maintain pluripotency.16 Another class, called ‘enhancer RNAs,’ was reported to work in cis to activate the expression of neighboring genes.15, 22 In contrast to microRNAs or other small ncRNAs, lincRNAs are not yet well classified and their general functions are still unknown.
Although the functions of lincRNAs are largely unknown, they have become an important factor in cancer biology. Several lincRNAs, including HOTAIR and ANRIL, were reported to be essential effectors in cancer.13, 23, 24 They regulate cancer-related gene expression both by epigenetic control and by interacting with chromatin-modifying proteins, such as EZH2, LSD1 and CBX7.11, 13, 24, 25 Several lincRNAs, including PCA3 and HOTAIR, are potential diagnostic or prognostic markers for cancer patients.13, 23, 26 Therefore, the discovery and characterization of cancer-related lincRNAs is important to both the biological and clinical fields in cancer research.
In this study we performed RNA-seq experiments comparing gastric cancer with normal tissues. Using our own RNA-seq data, as well as public DNA microarray data, we identified differentially expressed putative lincRNAs. We then examined their expression patterns, cancer-related phenotypes and effects on cancer-related molecules. Our results suggest that the lincRNAs that we identified in the present study have the potential to be lincRNA markers and therapeutic targets in gastric cancer.
Materials and methods
Tissue preparation and cell culture
Human gastric cancer samples and adjacent normal tissues were obtained from the Bio-Resource Center of the Asan Medical Center (Seoul, Korea) and Department of Pathology in Chungnam National University (Daejeon, Korea). All tissue samples were collected after obtaining informed consent under Institutional Review Board.
For the primary cell cultures, tissues were minced with scissors and digested for 3 h in minimal essential medium (Invitrogen, Carlsbad, CA, USA) containing 0.1 mg ml−1 type I collagenase (Sigma-Aldrich, St Louis, MO, USA). The isolated cells were washed with minimal essential medium and then with Dulbecco’s modified Eagle’s medium plus 10% fetal bovine serum (Lonza Group, Basel, Switzerland). The cells were then plated in bronchiolar epithelial growth medium or renal epithelial growth medium (Lonza Group) on collagen-coated dishes (Invitrogen) and were cultured at 37 °C in a humidified 5% CO2 incubator.
Gastric cancer cell lines were cultured in complete RPMI 1640 medium (WelGENE, Daegu, Korea). B16F1 mouse melanoma cell lines were cultured in Dulbecco’s modified Eagle’s medium (WelGENE). Cell lines were obtained from the Korean Cell Line Bank (http://cellbank.snu.ac.kr/index.html). All complete media contained 10% fetal bovine serum (WelGENE), 100 U ml−1 penicillin/streptomycin (Invitrogen) and 2 mM L-glutamine.
RNA isolation, cDNA synthesis and PCR experiments
Total RNA was isolated using either Trizol (Invitrogen) or RNeasy kit (QIAGEN, Valencia, CA, USA) according to the manufacturer’s instructions. The concentration of RNA was determined using a spectrophotometer and Experion RNA StdSens (BIO-RAD, Hercules, CA, USA), and the integrity of the RNA was verified using agarose gel electrophoresis. Using total RNA as a template, cDNAs were synthesized using iScript cDNA Synthesis Kits (BIO-RAD). Reverse-transcription PCR (RT-PCR) assays were performed using Novelzyme Taq Plus Premix (Noble Bio, Suwon, Korea). The quantitative real-time PCR (qRT-PCR) reactions with iQ SYBR Green Real-Time PCR Supermix (BIO-RAD) were performed on a CFX96 Real-Time PCR machine (C1000 Thermal Cycler, BIO-RAD) according to the following parameters: an initial denaturation step at 94 °C for 1 min, followed by 40 cycles of denaturation at 94 °C for 15 s and a final annealing/elongation step at 60 °C for 1 min. β-Actin was used as a housekeeping control gene for normalization. Expression levels were quantified using delta Ct (ΔCt). The RT-PCR and real-time qPCR primers were designed using either Primer3 software (http://frodo.wi.mit.edu/) or manually. All oligonucleotide primer sequences are listed in Supplementary Table 5.
RNA-seq experiment and data analysis
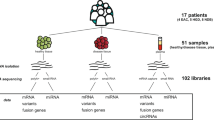
Poly(A)+RNA was selected from 3 μg total RNA using Sera-Mag oligo(dT) beads (Thermo Scientific, Lafayette, CA, USA), and paired-end next-generation sequencing libraries were prepared using Illumina-supplied universal adaptor oligos and PCR primers (Illumina, San Diego, CA, USA). Samples were sequenced on an Illumina Genome Analyzer II flow cell according to the manufacturers’ protocol. Seventy-six base pair paired-end reads were obtained.
TopHat (version 1.3.1; http://tophat.cbcb.umd.edu/) and Cufflinks (version 1.0.3; http://cufflinks.cbcb.umd.edu/) programs were used for short-read gapped alignment and ab initio assembly, respectively, to predict putative transcripts. When performing assembly with the Cufflinks program, we used one of two methods: with or without −G option (Supplementary Figure 1). We used the Affymetrix U133 Plus2 (affyU133Plus2, GPL570) gene model provided by the UCSC database (Supplementary Figure 1). For the with –G option, read counts in the affyU133Plus2 gene model were calculated based on the RPKM (reads per kilobase of exon per million fragments mapped) values provided by Cufflinks. For the without –G option, we divided the whole genome into 200-nucleotide bins and calculated the RPKM values using custom python scripts. Transcripts and bins sharing genomic positions with UCSC Known Genes were removed. Intergenic differentially expressed transcripts (iDETs) were selected by Student’s t-test between normal and cancer tissue/cell samples based on their RPKM values using the R program (http://www.r-project.org/) and Python programming.
Public microarray data analysis
Affymetrix U133 Plus 2 (GPL570) platform DNA microarray data about gastric cancer tissues were collected from the Gene Expression database of Normal and Tumor tissues (http://medical-genome.kribb.re.kr/GENT/) database. A total of 6154 probes on the GPL570 platform existed in intergenic regions. Collected microarray data were globally normalized with the MAS5 method using the affy package. iDETs were selected after evaluating significance using both the R program and Python programming.
For the survival analysis, we collected GPL570 platform DNA microarray data with survival data from Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/). Collected data sets were GSE6532, GSE9195, GSE20711, GSE21653, GSE31210, GSE37745, GSE2658, GSE19234, GSE18520, GSE19829, GSE30161, GSE7696, GSE16581, GSE31595, GSE10846, GSE11318, GSE23501, GSE12417 and GSE22762. Survival analysis was performed using R program.
Both the drawing of heatmaps and unsupervised hierarchical clustering were performed using MEV 4.0 program (http://www.tm4.org/). Read distribution drawing was performed using the UCSC genome browser (http://genome.ucsc.edu/), R and Python programming.
Overexpression and siRNA knockdown studies
The full-length clone of BM742401 was provided by 21C Human Gene Bank, Genome Research Center, KRIBB, Korea (http://genbank.kribb.re.kr) and inserted into a pcDNA3.1(+) expression vector. The insert sequence was confirmed by bidirectional sequencing. Cloned pcDNA3.1(+)–BM742401 were transfected into two gastric cancer cell lines, AGS and MKN-1, and one mouse melanoma cell line, B16F1, using Lipofectamine Plus (Invitrogen). The transformed cell lines were cultured and selected for using Geneticin (G418) for 2–3 weeks.
MKN-1 cells were plated and transfected with either 20 nM small interfering RNA (siRNA) oligos or non-targeting controls. Transfections were performed using Lipofectamine RNAiMAX Reagent (Invitrogen) in OptiMEM media. Knockdown was confirmed using RT-PCR at 48 h after transfection. The siRNAs for chr7_138 knockdown were designed by AsiDesigner (http://sysbio.kribb.re.kr:8080/AsiDesigner). The sequences were as follows: siRNA 1 sense 5′-CACUUGGUAGUGAAGACAU(AU)-3′; siRNA 1 antisense 5′-AUGUCUUCACUACCAAGUG(UU)-3′; siRNA 2 sense 5′-UUCUUACAGGCCUAACAUA(GC)-3′; siRNA 2 antisense 5′-UAUGUUAGGCCUGUAAGAA(UG)-3′.
Anchorage-independent growth, cell viability, migration and invasion assays
To evaluate anchorage-independent growth, suspensions of 1 × 103 cells were mixed with 0.4% agar (Sigma-Aldrich) in complete growth medium and seeded into six-well plates coated with 0.8% hardened agar. The plates were incubated at 37 °C for 20 days. Colonies were observed using light microscopy.
To evaluate cell viability, suspensions of 2 × 103 cells were seeded into 96-well plates and transfected with either 20 nM siRNA oligos or non-targeting controls. After 48 h at 37 °C in a humidified incubator, 20 μl of CellTiter-Blue Reagent (Promega, Madison, WI, USA) was added. After 2 h of incubation, the fluorescence intensity at 590 nm was measured.
Migration assays were performed using Transwell chambers (Corning, Corning, NY, USA) with 8 μm pore polycarbonate filters, and invasion assays were performed using BD BioCoat Matrigel Invasion Chambers (BD Biosciences, Bedford, MA, USA). Cells were suspended in serum-free media and counted. Cells were seeded into the upper chamber at a density of 2 × 104 for the migration assay and 1 × 105 for the invasion assay, and serum-containing media was placed into the lower chamber. After incubation for 24–48 h, cells that had penetrated the pores were stained with a staining solution (0.1% crystal violet in ethanol) and observed using a microscope.
Mouse in vivo metastasis (tail-vein injection) assay
Seven-week-old male C57BL/6 mice were used for an in vivo metastasis (tail-vein injection) assay. BM742401-overexpressing B16F1 cells were injected at a concentration of 5 × 106 cells in 200 μl phosphate-buffered saline into the tail veins of the mice. Mice were killed 3 weeks later. Their lungs were excised, fixed in formalin overnight, embedded in paraffin and hematoxylin and eosin stained.
Zymography assay
Proteins concentrated from a sample of cell supernatant were electrophoresed in 10% polyacrylamide gel containing 0.1% gelatin. SDS was removed from the gel by washing it with 2.5% Triton X-100. The gel was incubated overnight in reaction buffer (50 mM Tris (pH 7.5), 150 mM NaCl, 10 mM CaCl2, 0.02% NaN3, 2 μM ZnCl2 and 10 mM Triton X-100) and was subsequently stained with 0.5% Coomassie brilliant blue, followed by destaining.
Enzyme-linked immunosorbent assay
The concentration of MMP9 in the cell culture supernatant was determined by a Human MMP-9 ELISA Kit (RayBio, Norcross, GA, USA) according to the manufacturer’s instructions.
Accession numbers
All primary RNA-seq data are deposited in the Gene Expression Omnibus under accession number GSE41476.
Results
Identification of differentially expressed intergenic transcripts
We performed RNA-seq experiments to identify iDETs between gastric cancer and normal tissues/cells. We sequenced three primary cell culture samples from gastric cancer tissues, three gastric cancer cell lines and two normal tissue samples. Using the Illumina Genome Analyzer II platform, we obtained 353 182 315 sequence reads, among which 218 606 834 reads passed a filter of average Phred scores above 20. Using the TopHat program, we performed short-read gapped alignment. A total of 109 014 455 reads were mapped on the UCSC hg18 human genome (Supplementary Table 1). We performed ab initio assembly using the Cufflinks program to predict putative transcripts from the mapped reads.
When performing assembly and calculating normalized read counts, we used two methods (Supplementary Figure 1). First, we counted reads of putative transcripts within the UCSC affyU133Plus2 gene model. Second, we counted reads out of the gene model. In both cases, transcripts sharing a genomic position with UCSC Known Genes were removed. We performed Student’s t-test between normal and cancer tissue/cell samples, and selected 284 iDETs within and 143 iDETs out of the UCSC affyU133Plus2 gene model from RNA-seq data (Figure 1a and Supplementary Tables 2 and 3).

Screening of gastric cancer-related intergenic transcripts using RNA-seq and public microarray data. (a) Unsupervised hierarchical clustering of selected transcripts from RNA-seq data. Two hundred and eighty-four iDETs within the affyU133Plus2 gene model (left) and 143 200-nucleotide bins (right) out of the affyU133Plus2 gene model were selected. (b) Selection of iDETs using RNA-seq and public microarray data. Thirty-nine iDETs were selected by intersecting two lists of iDETs (top). The iDETs showed different expression patterns between cancer and normal tissue (bottom). (c) Read distribution of differentially expressed putative lincRNAs.
For transcripts within the affyU133Plus2 gene model, we took advantage of public microarray data to increase sample size when we selected iDETs. Using the Gene Expression database of Normal and Tumor tissues, we obtained a gene expression data of 57 gastric normal and 268 gastric tumor tissue samples produced using the Affymetrix U133. We selected 976 iDETs by performing Student’s t-test on the microarray data. We obtained 39 iDETs after intersecting the two lists of iDETs (Supplementary Table 4 and Figure 1b). We selected 31 iDETs after filtering out eight iDETs that were incongruent between RNA-seq and microarray data. These iDETs were supported by two platforms and a large number of gastric cancer samples.
To select iDETs for further studies, we applied more stringent filtering criteria: (1) high-expression levels; (2) similar expression patterns in other tissues; and (3) the existence of protein-coding genes near the iDETs (to test for cis or trans actions). One iDET within and the second iDET out of the affyU133Plus2 gene model were selected for further studies (Figure 1c). The first one was probed by 236118_at Affymetrix probe and located on chr18:18000855–18001676 genomic position (236118_at). The second one was located within chr7:138357000–138360000 genomic position (chr7_138). As shown in Figure 1c 236118_at was downregulated, whereas chr7_138 was upregulated in gastric cancer. The downregulation of 236118_at was observed in many cancer types (Supplementary Figure 2). Some known transcripts overlapped with 236118_at or chr7_138 as shown in the UCSC genome browser (Supplementary Figure 3). At genomic position 236118_at, we found two known transcripts: BM742401, which had an intron on chr18:18001268–18001562, and AK123079, which had no intron. Considering the reads distribution from the RNA-seq data and RT-PCR result, we determined that BM742401 was a major transcript at this genomic position (Supplementary Figures 3 and 4). At the genomic position of chr7_138, we found two representative known transcripts: BC020784 and AK098156. As we targeted iDETs out of the affyU133Plus2 gene model in this case, we selected AK098156 for further study (Supplementary Figures 3 and 5). Then, we characterized these two putative lincRNAs, BM742401 and AK098156.
Susceptibility of patients expressing the putative lincRNAs to gastric cancer
The two lincRNAs were previously known but were not well-characterized transcripts. We examined the expression of the two lincRNAs in seven gastric cell lines (monolayer cells, except SNU-620). As we expected, gastric cell lines expressed both BM742401 and AK098156 transcripts (Figure 2a).

Validation and survival analysis of putative lincRNAs, BM742401 and AK098156. Differential expression of the putative lincRNAs was validated using RT-PCR and real-time qPCR. (a) The expression of the lincRNAs in various gastric cancer cell lines. The lincRNAs were detected in various gastric cancer cell lines by RT-PCR. (b) Differential expression of BM742401 between tumor and normal tissues. (c) Differential expression of AK098156 between tumor and normal tissues. (d) Stage-specific expression pattern of BM742401. (e) Kaplan–Meier plot of gastric cancer patients’ survival based on differences in BM742401 expression. (f) Kaplan–Meier plot of stage III gastric cancer patients’ survival based on differences in BM742401 expression. Tumor, tumor tissue; NT, adjacent normal tissue.
We performed real-time qPCR on the two transcripts with 113 paired normal and tumor tissues from gastric cancer patients. The expression of BM742401 was significantly reduced in gastric tumor tissues (P=0.045; Figure 2b), whereas the expression of AK098156 was significantly increased in gastric tumor tissues (P=0.0014; Figure 2c). Moreover, BM742401 showed a stage III-specific expression pattern (Stage I: P=0.71; Stage II: P=0.66; Stage III: P=1.5 × 10−4; Stage IV: P=0.30; Figure 2d).
Using the real-time qPCR data and clinical information on the 113 gastric cancer patients, we performed a survival analysis (Figure 2e). For BM742401, we separated 113 patients into two groups based on the ΔCt value of −6.5 (median) in tumor tissue. Lower expression group showed poorer survival than higher expression group (Figure 2e; P=4.8 × 10−3 by log-rank test). We tested the value of BM742401as, a prognostic marker for gastric cancer, using a Cox proportional hazards model with variants such as tumor stages (Table 1). BM742401 was less significant than conventional prognosis markers such as tumor stage. However, when we restricted Cox analysis to stage III patients (n=35), BM742401 expression level was more prognostic than grouping by stage IIIA and IIIB (Table 2). Moreover, low-expression (ΔCt<−6.5) group also had poorer survival than high-expression group among stage III patients (Figure 2f; P=0.062). The expression level of AK098156 was not prognostic on gastric cancer patients’ survival (data not shown).
BM742401 expression was downregulated in many cancer types (Supplementary Figure 2). We tested whether BM742401 expression was prognostic in other cancer patients. From public gene expression data sets, we found that the low-expression groups had a tendency to show poorer survival than the high-expression groups in several solid cancers, such as breast, lung, myeloma and melanoma (Supplementary Figure 6). Moreover, downregulation of BM742401 was significantly associated with poor recurrence- and metastasis-free survival in GSE9195 breast cancer data set. We found no public microarray data probing AK098156. As BM742401 was prognostic in many cancer types, we decided to study BM742401 more than AK098156 (Supplementary Figure 7).
Regulation of cancer metastasis by BM742701 lincRNA
As BM742401 was expressed at low levels in most monolayer gastric cancer cell lines (Figure 2a), we overexpressed BM742401 in AGS and MKN-1 cells, and observed its effects in vitro. We first confirmed the overexpression of BM742401 in both cell lines by RT-PCR (Figures 3a and b top). BM742401 overexpression did not influence cell viability and colony formation of gastric cancer cells (Supplementary Figure 8), but it significantly decreased migration and invasion ability (Figures 3a and b, and Supplementary Figures 9 and 10). As BM742401 was downregulated in many cancer types (Supplementary Figure 2), we performed the same assays in B16F1 mouse melanoma cell line (Figure 3c). BM742401 overexpression also significantly decreased migration and invasion ability of B16F1 cell line. Thus, we found that BM742401 regulated specifically metastasis-related phenotypes.

Metastasis-related in vitro phenotype assays for BM742401. Using stably BM742401-overexpressed cancer cell lines, migration and invasion assays were performed. (a) Assays for MKN-1 cell line. (b) Assays for AGS cell line. (c) Assays for B16F1 cell line.
As BM742401 decreased migration and invasion of cancer cells in vitro, we further examined whether it could influence cancer metastasis in vivo. We then injected the control and BM742401-overexpressing B16F1 cells into the tail veins of mice, and after 3 weeks killed the mice and isolated their lungs. Black metastatic foci were observed on and inside their lungs in both types of mice, but BM742401 overexpression significantly reduced the size and number of foci (Figure 4a). Hematoxylin and eosin staining of the paraffin-embedded lung tissues also allowed us to observe a decrease in the size and number of the metastatic foci (Figure 4b). We concluded that BM742401 overexpression decreased cancer metastasis by regulating the migration and invasion of cancer cells.

Metastasis-related in vivo phenotype assay after BM742401 overexpression. (a) Lungs from mice injected with BM742401-overexpressing B16F1 or control cells into their tail veins. Black foci are metastasized B16F1 cells. (b) Hematoxylin and eosin staining of the separated and paraffin-embedded lungs.
Regulation of extracellular MMP9 by BM742401
We investigated how BM742401 regulated the migration and invasion of cancer cells. Matrix metalloproteinases (MMPs) are proteins that regulate cancer cell invasiveness, and MMP2 and MMP9 are known as representative gelatinases of the extracellular matrix.27, 28 At first, we measured MMP activity using zymography assay with culture supernatants obtained from control and BM742401-overexpressing cells (Figure 5a). BM742401 overexpression decreased the activity of the ∼95 kDa band, the size of which corresponds to that of MMP9. The activity of the lower band that may represent MMP2 (around 70 kDa) was not changed by BM742401 overexpression. Therefore, we measured the MMP9 concentration using an MMP9 enzyme-linked immunosorbent assay kit and found that extracellular MMP9 was indeed reduced by BM742401 overexpression (Figure 5b). We tested whether the intracellular MMP9 expression was inhibited by BM742401 overexpression using RT-PCR, real-time qPCR, immunoblot assay and enzyme-linked immunosorbent assay (Supplementary Figure 11). But, BM742401 did not influence intracellular MMP9 expression. Thus, we concluded that BM742401 inhibited cancer metastasis by regulating MMP9 secretion.

Inhibition of extracellular MMP9 by BM742401 overexpression. (a) Extracellular enzyme activity of MMPs (zymography assay). (b) Extracellular MMP9 concentration (enzyme-linked immunosorbent assay).
Discussion
Several lincRNAs have become important effectors and diagnostic/prognostic markers in various cancers.13, 23, 24, 26, 29 One well-known lincRNA, H19, was reported to have a role in gastric cancer.29 We found 31 novel lincRNAs that were differentially expressed in gastric cancer using our own RNA-seq, as well as public DNA microarray data. Two of these lincRNAs regulated either proliferation or metastasis-related phenotypes in gastric cancer cells. Moreover, one of them, BM742401, influenced both the survival rate of cancer patients and the levels of a metastasis-related molecule.
Each of the two sets of transcriptomics data (our own RNA-seq data and the public microarray data) had its own advantages and disadvantages. RNA-seq data provide the expression patterns of whole intra- and intergenic transcriptomes at a single-nucleotide resolution, but the number of samples was too small for statistically reliable results. The public DNA microarray data, on the other hand, had a sufficient number of samples with additional clinical information, including survival data, but the probes on the microarray represented only predefined transcripts and had low resolution when compared with the RNA-seq. As those two data sets were complementary to one another, we selected the intersection of iDETs from both data sets.
One of the challenges in studying lincRNAs is that little information is available for intergenic transcripts. Fortunately, our candidates had several known sequences in expressed sequence tag, Gene Bank and other databases; hence, we could do further studies based on that information. For further selection, we considered three criteria for the putative lincRNAs and finally selected two candidates: 236118_at and chr7_138. Several known transcripts existed at the same genomic position as the two candidates. Microarray probes cannot separate transcripts at the same genomic position if they are not specially designed. If we had used only microarray data for the selection, we could not have selected one representative transcript. The RNA-seq data showed us which transcript was the predominant one. Considering the distribution of reads, we selected one representative transcript. In our opinion, it is another merit of the RNA-seq platform to study intergenic transcripts.
Downregulation of BM742401 significantly reduced the survival of gastric cancer patients, but the reduction in survival was less significant than tumor stage. However, the expression of BM742401 separated poor survival of stage III patients more efficiently than grouping into stage IIIA and IIB. Therefore, we think that BM742401 could be a putative subtype marker for the prognosis of stage III gastric cancer patient survival.
For the BM742401 lincRNA that was within affyU133Plus2 gene model, we could use public microarray data with survival data. Downregulation of BM742401 was associated with poor survival of various solid cancer patients in public microarray data. Especially, it was associated with reduced recurrence- and metastasis-free survival in breast cancer patients. Thus, we supposed that BM742401 would regulate metastasis-related phenotypes.
One question about our putative lincRNAs was whether they were ncRNAs or protein-coding genes. We have two evidences indicating that our putative lincRNAs are not protein-coding genes: first, when we predicted open reading frames of our putative lincRNAs using gene prediction programs, such as GeneScan (http://genes.mit.edu/GENSCAN.html) and FGENESH (http://www.softberry.com/), we found no suitable open reading frames. Second, when we compared sequencing data with the reference genome sequence, we found that short-tandem repeats existed in both sequences. If they had been translated based on the triplet codon, it would have caused a frameshift and the translated protein would have undergone abnormal folding. Hence, we concluded that they were not protein-coding genes.
One controversial issue in lincRNA study is whether it works in cis or trans. We tested whether overexpression of BM742401 influenced the expression of neighbor genes, such as GATA6, but found that it did not change the expression of neighbor genes (data not shown). Thus, we concluded that BM742401 worked in trans or did not affect transcription.
The effect of BM742401 overexpression was small compared with the effects of protein-coding gene overexpression. For example, overexpression of BM742401 only reduced 20∼40% of cancer cell invasion and only ∼40% of extracellular MMP9. We thought that BM742401 would not be an effector molecule in and of itself but that it would be a helper, or cofactor, of other significant effectors. Although we tried to find molecules that interact with BM742401 using microarray, chromatin immunoprecipitation, biotinylated RNA pull-down and mass spectroscopy, we could not find any effector molecules that interact directly with BM742401 (data not shown).
In spite of its small effect size, BM742401 showed significant and specific influence over metastasis-related phenotypes, but not proliferation-related phenotypes. Considering the association of BM742401 with survival rate and its specific influence on metastasis-related phenotypes, we suggest that BM742401 is a potential specific lincRNA marker and therapeutic target in late-stage gastric cancer patients.
Accession codes
References
Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009; 458: 223–227.
Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci USA 2009; 106: 11667–11672.
Bertone P, Stolc V, Royce TE, Rozowsky JS, Urban AE, Zhu X et al. Global identification of human transcribed sequences with genome tiling arrays. Science 2004; 306: 2242–2246.
Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007; 316: 1484–1488.
Metzker ML . Sequencing technologies - the next generation. Nat Rev Genet 2010; 11: 31–46.
Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol 2010; 28: 503–510.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010; 28: 511–515.
Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT . Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 2008; 322: 750–756.
Leighton PA, Ingram RS, Eggenschwiler J, Efstratiadis A, Tilghman SM . Disruption of imprinting caused by deletion of the H19 gene region in mice. Nature 1995; 375: 34–39.
Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J et al. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell 2008; 32: 232–246.
Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007; 129: 1311–1323.
Mercer TR, Dinger ME, Mattick JS . Long non-coding RNAs: insights into functions. Nat Rev Genet 2009; 10: 155–159.
Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010; 464: 1071–1076.
Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010; 142: 409–419.
Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G et al. Long noncoding RNAs with enhancer-like function in human cells. Cell 2010; 143: 46–58.
Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011; 477: 295–300.
Hung T, Wang Y, Lin MF, Koegel AK, Kotake Y, Grant GD et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet 2011; 43: 621–629.
Loewer S, Cabili MN, Guttman M, Loh YH, Thomas K, Park IH et al. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat Genet 2010; 42: 1113–1117.
Ponting CP, Oliver PL, Reik W . Evolution and functions of long noncoding RNAs. Cell 2009; 136: 629–641.
Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R et al. The air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 2008; 322: 1717–1720.
Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K et al. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell 2010; 40: 939–953.
Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 2011; 472: 120–124.
Kim K, Jutooru I, Chadalapaka G, Johnson G, Frank J, Burghardt R et al. HOTAIR is a negative prognostic factor and exhibits pro-oncogenic activity in pancreatic cancer. Oncogene 2012; 32: 1616–1625.
Yap KL, Li S, Munoz-Cabello AM, Raguz S, Zeng L, Mujtaba S et al. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell 2010; 38: 662–674.
Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010; 329: 689–693.
de Kok JB, Verhaegh GW, Roelofs RW, Hessels D, Kiemeney LA, Aalders TW et al. DD3(PCA3), a very sensitive and specific marker to detect prostate tumors. Cancer Res 2002; 62: 2695–2698.
Westermarck J, Kahari VM . Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J 1999; 13: 781–792.
Vihinen P, Kahari VM . Matrix metalloproteinases in cancer: prognostic markers and therapeutic targets. Int J Cancer 2002; 99: 157–166.
Yang F, Bi J, Xue X, Zheng L, Zhi K, Hua J et al. Up-regulated long non-coding RNA H19 contributes to proliferation of gastric cancer cells. FEBS J 2012; 279: 3159–3165.
Acknowledgements
This was work supported by grants from stem cell (2012M3A9B4027954) and genomics (2012M3A9D1054670) program of the National Research Foundation of Korea, funded by the Ministry of Education, Science and Technology (MOEST), and from the KRIBB Research Initiative.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Experimental & Molecular Medicine website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Park, SM., Park, SJ., Kim, HJ. et al. A known expressed sequence tag, BM742401, is a potent lincRNA inhibiting cancer metastasis. Exp Mol Med 45, e31 (2013). https://doi.org/10.1038/emm.2013.59
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2013.59
Keywords
This article is cited by
-
Epigenetic silencing of long non-coding RNA BM742401 in multiple myeloma: impact on prognosis and myeloma dissemination
Cancer Cell International (2020)
-
Emerging roles of long non-coding RNAs in cancer
Journal of Biosciences (2019)
-
A RNA-Sequencing approach for the identification of novel long non-coding RNA biomarkers in colorectal cancer
Scientific Reports (2018)
-
Epigenetic roles in the malignant transformation of gastric mucosal cells
Cellular and Molecular Life Sciences (2016)
-
Long non-coding RNAs in cancer invasion and metastasis
Modern Pathology (2015)
