
Abstract
Alzheimer’s disease (AD) is the most common cause of age-related dementia. The neuropathological hallmarks of AD include extracellular deposition of amyloid-β peptides and neurofibrillary tangles that lead to intracellular hyperphosphorylated tau in the brain. Soluble amyloid-β oligomers are the primary pathogenic factor leading to cognitive impairment in AD. Neural stem cells (NSCs) are able to self-renew and give rise to multiple neural cell lineages in both developing and adult central nervous systems. To explore the relationship between AD-related pathology and the behaviors of NSCs that enable neuroregeneration, a number of studies have used animal and in vitro models to investigate the role of amyloid-β on NSCs derived from various brain regions at different developmental stages. However, the Aβ effects on NSCs remain poorly understood because of conflicting results. To investigate the effects of amyloid-β oligomers on human NSCs, we established amyloid precursor protein Swedish mutant-expressing cells and identified cell-derived amyloid-β oligomers in the culture media. Human NSCs were isolated from an aborted fetal telencephalon at 13 weeks of gestation and expanded in culture as neurospheres. Human NSCs exposure to cell-derived amyloid-β oligomers decreased dividing potential resulting from senescence through telomere attrition, impaired neurogenesis and promoted gliogenesis, and attenuated mobility. These amyloid-β oligomers modulated the proliferation, differentiation and migration patterns of human NSCs via a glycogen synthase kinase-3β-mediated signaling pathway. These findings contribute to the development of human NSC-based therapy for AD by elucidating the effects of Aβ oligomers on human NSCs.
Similar content being viewed by others

Introduction
Neural stem cells (NSCs) generated from developing and adult mammalian brains are defined by their ability to self-renew, give rise to glial and neuronal cell lineages throughout the neuraxis and reconstruct developing or damaged central nervous system regions.1, 2, 3, 4 Studies of NSCs isolated from human fetal brains have advanced clinical approaches providing replacement and protection of neural cells against degenerated or injured stimuli5, 6, 7, 8, 9, 10 and have facilitated an increased understanding of human brain development.1
Alzheimer’s disease (AD) is a dire neurodegenerative disease that frequently occurs in elderly adults. AD neuropathologically manifests with extracellular deposition of amyloid-β (Aβ), intraneuronal accumulation of hyperphosphorylated tau and inflammation in the brain. The accompanying synaptic dysfunction and neuronal loss leads to severe cognitive deficits.11, 12, 13 Although the primary cause of AD has not yet been established, substantial evidence suggests that Aβ cleaved from amyloid precursor protein (APP) by β- and γ-secretase contributes to the development of AD, and soluble oligomeric species of Aβ, rather than Aβ plaques, induce synaptic failure, neuronal toxicity and memory decline in AD-like animal models.14, 15, 16 Interestingly, cell- and AD brain-derived low-n oligomers of Aβ have biologically detrimental effects on neurons,17, 18, 19, 20 even though these oligomers are smaller than synthetic Aβ oligomers.21, 22 In addition, Aβ oligomers and peptides activate glycogen synthase kinase-3β (GSK-3β), whose activity is inversely regulated by the Wnt/β-catenin signaling pathway23, 24 involved in the phosphorylation of tau22, 25 and related to APP processing,26 in AD brains and rat hippocampal neurons.
A number of previous studies have investigated how NSCs are modulated in the brains of AD patients by studying diverse Aβ species and using different animal and cell culture models. Based on these investigations, each Aβ species may play a role in regulating proliferation, differentiation and migration of NSCs, although the results of these studies are inconsistent with reports that Aβ either decreases or increases dividing potential, prevents or induces neurogenesis and inhibits the movement of NSCs in adult transgenic animals or in vitro.23, 27, 28, 29, 30, 31, 32, 33, 34, 35 Furthermore, some studies have reported hippocampal neurogenesis in AD patients.31, 36, 37 However, neurogenesis in the dentate gyri of AD patients may be difficult to interpret because of differences in symptom levels, age and post-mortem delays in the processing of patient samples. Thus, the Aβ effects on NSCs remain poorly understood.
In the present study, we demonstrate that naturally secreted Aβ oligomers derived from APP Swedish mutant (APPsw)-expressing cells regulate the proliferation, differentiation and migration patterns of human fetal brain-derived NSCs via a GSK-3β-mediated signaling pathway. These findings contribute to the development of human NSC-based therapy for AD by elucidating the effects of Aβ oligomers on the properties of human NSCs.
Materials and methods
Culture of human NSCs
Human fetal tissue from a cadaver at 13 weeks of gestation was obtained with full parental consent and approval of the research ethics committee of Yonsei University College of Medicine, Seoul, Korea. All procedures conformed to the guidelines of both the National Institutes of Health and the Korean Government. The freshly dissected telencephalic tissue was transferred to the Good Manufacturing Practice facility, and after dissociation in trypsin (0.1% for 30 min, Sigma, St Louis, MO, USA), seeded onto tissue-culture-treated 100-mm plates (Corning, Tewksbury, MA, USA) at a density of 400 000 cells per ml in serum-free N2 growth medium, which consisted of a 1:1 mixture of Dulbecco’s modified Eagle’s medium and Ham’s F12 (DMEM/F12; Gibco, Grand Island, NY, USA), supplemented with 1 × penicillin/streptomycin (P/S; Gibco) and N2 formulation (1% v/v; Gibco). Mitogenic stimulation was achieved by adding 20 ng ml−1 fibroblast growth factor-2 (R&D Systems, Minneapolis, MN, USA), and 10 ng ml−1 leukemia inhibitory factor (Sigma). Heparin (8 μg ml−1; Sigma) was added to stabilize fibroblast growth factor-2 activity. All cultures were maintained in a humidified incubator at 37 °C and 5% CO2, and half of the growth medium was replenished every 3–4 days. Proliferating single cells in culture gave rise to free-floating neurospheres during the first 2–5 days of growth. Cells were passaged every 7–8 days by dissociation of bulk neurospheres with 0.05% trypsin/EDTA (Gibco).
Viral vector transduction
To express human APP695sw-green fluorescent protein (APPsw-GFP) and GFP, recombinant lentiviral particles were prepared. The 293T cells were cultured in DMEM (Gibco) containing 10% fetal bovine serum (Gibco) and 1 × P/S. Subconfluent 293T cells in DMEM were co-transfected with 20 μg pWPI transfer vector containing APPsw-GFP, 15 μg psPAX2 packaging construct and 6 μg pMD2G envelope plasmid by calcium phosphate precipitation. The lentiviral vectors were kindly donated by the Trono laboratory (Addgene, Cambridge, MA, USA). After 3 h, fresh DMEM containing 5% fetal bovine serum and 1 × P/S was added. After 2 days, the culture media containing lentiviral particles were harvested, centrifuged at 3000 r.p.m. for 5 min to remove cell debris and concentrated by ultracentrifugation at 26 000 r.p.m. under a sucrose cushion. The viral titer was determined as transducing units using flow cytometry, guided by the protocol of the Trono lab (http://tronolab.epfl.ch/). Lentiviral particles encoding APPsw-GFP and GFP were respectively transduced into SK-N-MC cells (human neuroblastoma cells) or human NSCs with a multiplicity of infection of 1, and these cells were maintained for 1 month and used for subsequent experiments.
Preparation of conditioned media
SK-N-MC cells untransduced or transduced with lenti-APPsw-GFP or lenti-GFP were cultured in DMEM containing 10% fetal bovine serum and 1 × P/S. To prepare conditioned media (CM), these cells were seeded at a density of 6 × 106 on 100 mm culture dishes in 10 ml N2 media and incubated for 3 days. At the end of this period, media were harvested and cleared by centrifugation at 3000 r.p.m. for 5 min. The CM was divided into aliquots and stored at −70 °C until use.
Neurotoxicity of CM derived from APPsw-expressing SK-N-MC cells
Three-month-old-C57BL6/J mouse brains were extracted, embedded in 3% agarose and coronally cut on a vibratome (Motorized Advance Vibroslice MA752; Campden Instrument, Lafayette, IN, USA) in cold phosphate-buffered saline (PBS). Four to five 500-μm-thick slices throughout the cortex were cultured for 1 day and slices incubated in CM from untransduced SK-N-MC cells (SK-CM), CM from lenti-GFP-transduced SK-N-MC cells (GFP-CM), CM from lenti-APPsw-GFP-transduced SK-N-MC cells (APP-CM) and Aβ-immunodepleted or isotype-matched IgG-treated APP-CM. Mouse anti-Aβ1-16 (6E10 2 μg; Covance, Princeton, NJ, USA) or ChromPure mouse IgG (2 μg; Jackson ImmunoResearch, West Grove, PA, USA) were used for immunoprecipitation using Dynabeads ProteinG (Invitrogen, Grand Island, NY, USA) as described below. After 48 h, brain slices were washed with cold PBS, fixed in cold 4% paraformaldehyde (PFA) in PBS at 4 °C overnight, and subsequently transferred into a 30% sucrose solution at 4 °C until embedding. The cryoprotected brain slices in OCT compound (Sakura Finetek, Torrance, CA, USA) were cut at a thickness of 20 μm on slides using a Leica CM1850 cryostat (Wetzlar, Germany) and stored at −20 °C. The sections were incubated with freshly prepared permeabilization solution (0.1% Triton X-100 in 0.1% sodium citrate) for 10 min at 4 °C. After washing with PBS, sections were treated with the terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) reaction mixture (a 1:9 mixture of enzyme solutions: label solutions), processed using an in situ Cell Death Detection Kit (Roche Applied Science, Mannheim, Germany) for 1 h at 37 °C and washed with PBS. These sections were blocked with 10% normal donkey serum (Jackson ImmunoResearch) and 3% bovine serum albumin (BSA; Sigma) in PBS containing 0.3% Triton X-100 and incubated at 4 °C overnight with mouse anti-NeuN (1:100; Millipore, Billerica, MA, USA). Appropriate secondary antibodies conjugated with Dylight 594 (Jackson ImmunoResearch) were applied for 70 min at 37 °C followed by PBS washes, and then mounted with VECTASHIELD Mounting Medium (Vector, Burlingame, CA, USA). TUNEL-positive and NeuN-positive cells were observed using an Olympus BX51 microscope (Tokyo, Japan). PC12 cells were cultured in RPMI Media 1640 (Gibco) supplemented with 5% fetal bovine serum, 10% horse serum (Gibco) and 1 × P/S. Single PC12 cells dissociated with 0.05% trypsin/EDTA were seeded at a density of 5 × 105 cells per well on 12-well plates (Corning) in the growth medium. One day later, culture media were fully replaced with N2 media, SK-CM or APP-CM, and incubated for 36 h. Cell viability was measured using the Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s manual. Briefly, at the end of the incubation, CCK-8 solution was added to each well and incubated for 3 h at 37 °C. Media harvested from each well were transferred into a 96-well plate (Corning), and optical density measured at a wavelength of 450 nm.
Measurement of human NSC proliferation
Cell proliferation was estimated by 5′-bromo-2′-deoxyuridine (BrdU) incorporation. Human NSCs were seeded onto six-well plates at a density of 1 × 106 cells per well in N2 media to arrest the cell cycle. Each day, half of the culture media was replaced with CM containing mitogens and then cultured for 3 days. Neurospheres derived from these cells were dissociated as single cells, fixed in 70% ethanol, permeabilized using 2 N HCl and labeled with mouse anti-BrdU antibodies (Sigma) and fluorescein-conjugated anti-mouse IgG antibodies (Vector). The percentage of cells in S phase in the same samples was determined via propidium iodide staining. BrdU-immunolabeled cells and DNA content were analyzed by flow cytometry.
The TUNEL assay was performed using the In Situ Cell Death Detection Kit. Dissociated single cells mentioned above were fixed with 4% PFA in PBS for 1 h at room temperature. The samples were washed with PBS and then permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate buffer for 15 min at 4 °C. After 3 washes, cells were resuspended in TUNEL reaction mixture and incubated for 60 min at 37 °C. TUNEL-positive cells were quantified by flow cytometry, on a BD FACSCalibur (BD Biosciences, San Jose, CA, USA), reading 10 000–20 000 cells per condition. Cell distribution and staining with each dye were estimated using BD CellQuest, and images were obtained using either BD CellQuest or FlowJo 9.3.3 (Tree Star, Ashland, OR, USA).
Senescence-associated β-galactosidase activity assay
As described above, proliferating human NSCs treated with SK-CM or APP-CM for 3 days were collected, washed and resuspended in either 0.1 M citrate buffer (pH 4.5) or 0.1 M phosphate buffer (pH 6.0). Cells were lysed by freezing and thawing, and lysates centrifuged at 12 000 × g for 7 min. The supernatant was mixed with chlorophenol red-β-D-galactopyranoside (2.2 μg μl−1; Sigma) and 10 mM MgCl2. A heat inactivation control was prepared for 3 min at 65 °C. After an overnight incubation at 37 °C, 1.5 volumes of 1 M Na2CO3 were added to stop the reaction, each mixed sample was transferred to a 96-well plate; absorbance at 570 nm was then measured.
Telomere length
Quantitative real-time PCR amplification of the telomere sequence was performed as described previously.38 The relative telomere length (telomere (T)/single copy gene (S) ratio) was examined as the ratio of repeats of telomeres to the single copy gene 36B4, and calculated using the following formula: telomere length=2(−ΔCp) where ΔCp=CpTelomere−Cp36B4.39 PCR was performed with 30 ng genomic DNA, SYBR Green master mix (Roche Applied Science) and 200 nM primers in the LightCycler 480 System (Roche Applied Science) as follows: 95 °C for 2 min, 45 cycles of 95 °C for 10 s, 57 °C for 20 s and 72 °C for 60 s in a reaction volume of 20 μl in 96-well plates according to the manufacturer’s protocol. The following primer pairs were used: 5′-CGGTTTGTTTGGGTTTGGGTTTGGGTTTGGGTTTGGGTT-3′ and 5′-GGCTTGCCTTACCCTTACCCTTACCCTTACCCTTACCCT-3′ for telomeres (200 nM); 5′-CAGCAAGTGGGAAGGTGTAATCC-3′ and 5′-CCCATTCTATCATCAACGGGTACAA-3′ for 36B4 (300 nM).
Immunocytochemistry
A single-cell suspension generated from neurospheres was plated onto poly-L-lysine (10 μg ml−1; Sigma)-coated eight-well chamber slides (Nunc, Rochester, NY, USA) in N2 media. Each day, half of the culture media was replaced with SK-CM or APP-CM. On the sixth day, cells were fixed with 4% PFA in PBS for 10 min and rinsed three times with PBS. The cells were blocked with 3% BSA and 10% normal horse serum (Vector) in PBS containing 0.01% or 0.2% Triton X-100 at room temperature for 1 h and then incubated with primary antibodies: rabbit anti-nestin (1:200; Millipore), rabbit anti-GFAP (1:1500; DAKO, Glostrup, Denmark), rabbit anti-neuronal class III β-tubulin (Tuj1, 1:500; Covance), mouse anti-S100β (1:1000; Sigma), rabbit anti-galactocerebroside (GalC, 1:100; Sigma) and mouse anti-O4 (1:100; Millipore) at 4 °C overnight. After washing with PBS, slides were labeled with either fluorescein- or Texas red-conjugated anti-mouse or rabbit IgG antibodies (1:200; Vector). The percentage of immunoreactive cells was calculated using images acquired by an Olympus BX51 microscope. Three to five random fields were selected for each treatment, and cell ratio counts were performed on fields counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Vector).
Transplantation of human NSCs into NSE/APPsw transgenic mice
Heterogeneous NSE/APPsw transgenic mice harboring APPsw under the neuron-specific enolase (NSE) promoter40 were bred with background-matched C57BL/6J mice. Genotyping PCR was performed using genomic DNA isolated from newborn mouse tails at 3–4 weeks of age using the following primer pairs; 5′-CTGAGTCTGCAGTCCTCGA-3′ and 5′-CTCTTCTCACTGCATGTCTC-3′ for NSE-APPsw, and 5′-CTAGGCCACAGAATTGAAAGATCT-3′ and 5′-GTAGGTGGAAATTCTACATCATCC-3′ as an internal positive control. All animal studies were performed according to approved protocols of the institutional animal care and use committee of the Yonsei University College of Medicine.
A cell suspension of ∼3.2 × 105 BrdU-incorporating, lentiviral-GFP-transduced or untransduced human NSCs in 4 μl H-H buffer (1 × Hank’s balanced salt solution, 10 mM HEPES, pH 7.4; Invitrogen) was administered into the bilateral molecular layer of the dentate gyrus (A/P, −2.54 mm; L, ±2.5 mm from bregma; D/V, −2.25 mm from the dura mater) of 13-month-old NSE/APPsw transgenic and age-matched wild-type mice. Cyclosporine A (10 mg kg−1) was intraperitoneally injected daily beginning 1 day before transplantation. At 6 weeks after transplantation, these mice were transcardially perfused with cold PBS followed by cold 4% PFA in 0.1 M PIPES buffer (pH 6.9). The brains were carefully extracted, fixed in 4% PFA in 0.1 M PIPES buffer at 4 °C overnight and subsequently dehydrated in a 30% sucrose solution at 4 °C overnight. Brains were then cryoprotected in OCT compound, sectioned to an 18 μm thickness using a freezing cryostat (Leica) and stored at −20 °C. For immunohistochemistry, coronal brain sections were washed twice with PBS, and then blocked with 10% normal donkey serum and 3% BSA in PBS containing 0.3% Triton X-100. BrdU staining required a 30-min pretreatment step in 2 N HCl at 37 °C. Primary antibodies were incubated at 4 °C overnight, appropriate secondary antibodies conjugated with either Dylight488 or Dylight594 (Jackson ImmunoResearch) were applied for 70 min at 37 °C followed by three PBS washes and then mounted. The following primary antibodies were used to identify donor-derived cells: anti-BrdU-fluorescein (1:20; Roche Applied Science), rabbit anti-GFP (1:200; Invitrogen) and mouse anti-human cytoplasm (SC121, 1:500; StemCells, Newark, CA, USA); anti-nestin, Tuj1, glial fibrillary acidic protein (GFAP) and rabbit anti-olig2 (1:500; Millipore) were used to examine the fate of grafted human NSCs. Immunolabeled cells were observed and measured using an Olympus BX51 microscope and a Zeiss LSM 700 confocal microscope (Oberkochen, Germany).
Migration assay
Ten to fifteen neurospheres formed from singly dissociated human NSCs for 5 days were plated on poly-L-lysine-coated 12-well plates and maintained in N2 media for 1 day. The culture was refreshed daily with half SK-CM, GFP-CM or APP-CM for 3 days. To determine the migration of human NSCs from the neurospheres, cells were fixed on the fourth day and stained as described above. Areas of DAPI-positive migrating cells were measured from each neurospheres to the furthest migrating cell. Alternatively, 2.2 × 105 SK-N-MC cells transduced or not transduced with lenti-APPsw-GFP were seeded onto poly-L-lysine-coated 24-well plates (lower chamber). The bottom of the 3.0-μm porous insert (upper chamber) of transwell permeable supports (Corning) was coated with laminin and overlaid onto the plates. Dissociated human NSCs at a density of 1 × 105 were added into the upper chambers. Every day for 3 days, human NSCs that migrated through the pores and attached under the upper chamber were counted using DAPI staining. Images were acquired with an Olympus BX51 microscope and a Zeiss LSM 710 confocal microscope, and analyzed using the NIH ImageJ software (Bethesda, MD, USA).
Immunodepletion and pharmacological treatments
The 6E10 antibodies (5 μg ml−1) were added to APP-CM for 1 h at 37 °C before treatment, and another sample of APP-CM was incubated with ChromPure mouse IgG (5 μg ml−1) as an isotype control under the same conditions. TDZD-8 (Sigma) as an inhibitor of GSK-3β was used at 12.5 μM in CM before treatment. Control experiments were performed by adding an equal volume of the dimethyl sulfoxide (DMSO; Sigma) vehicle used to dissolve TDZD-8.
Western blot and enzyme-linked immunosorbent assay
To identify the species of secreted Aβ in APP-CM, 20-μl Dynabeads ProteinG (Invitrogen) were incubated with 2 μg mouse monoclonal anti-6E10 in PBS containing 0.02% Tween-20 at room temperature for 30 min. SK-CM and APP-CM were respectively added to 6E10-coated Dynabeads and incubated at 4 °C overnight. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis was then performed with proteins eluted from the Dynabeads complexes. To assess differences in secreted APP between SK-CM and APP-CM, CM was concentrated 10-fold using Amicon Ultra-0.5 centrifugal filter devices (Millipore) in accordance with the manufacturer’s guidelines. These concentrated CM were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis as described below.
SK-N-MC cells, lenti-APPsw-GFP or lenti-GFP transduced SK-N-MC cells and CM-treated human NSCs under the proliferating or differentiation conditions were lysed with M-PER Mammalian Protein Extraction Reagent (Thermo, Rockford, IL, USA) supplemented with Halt Protease and Phosphatase Inhibitor (Thermo). Protein concentrations were determined using the Pierce 660 nm Protein Assay (Thermo) and BSA as a standard. Proteins were resolved on a 4–15% Mini-PROTEAN TGX gel (Bio-Rad, Hercules, CA, USA) or 16.5% tricine gel41 under reducing conditions, and transferred to a nitrocellulose membrane. The membrane was incubated in 5% nonfat milk or 3% BSA in Tris-buffered saline containing 0.1% Tween-20 for 1 h. After an overnight incubation at 4 °C with primary antibodies, the membrane was washed in Tris-buffered saline containing 0.1% Tween-20 and incubated at room temperature with peroxidase-conjugated anti-mouse or rabbit antibodies (Jackson ImmunoResearch) for 1 h. The following primary antibodies used in a 1:1000 dilution: anti-6E10, mouse anti-APP (22C11; Millipore), mouse anti-proliferating cell nuclear antigen (PCNA; Cell Signaling, Danvers, MA, USA), mouse anti-S100β (Sigma), mouse anti-polysialic acid-NCAM (PSA-NCAM; Millipore), rabbit anti-myelin basic protein (MBP; Dako), rabbit anti-β-catenin (Cell Signaling), mouse anti-GSK3β (Santa Cruz Biotechnology, Dallas, TX, USA), rabbit anti-phospho-GSK3β (Cell Signaling), rabbit anti-Stat3 (Cell Signaling), rabbit anti-phospho-Stat3 (Cell Signaling) and β-actin (Sigma). The blots were washed in Tris-buffered saline containing 0.1% Tween-20, briefly treated with SuperSignal West Pico Chemiluminescent Substrate (Thermo) and processed in an LAS 4000 mini (GE Healthcare, Uppsala, Sweden) to detect immunolabeled bands. The band intensity of proteins of interest was arbitrarily estimated as a ratio to β-actin using a Multi Gauge V3 (Fujifilm, Tokyo, Japan). The enzyme-linked immunosorbent assay measurements of Aβ40 and Aβ42 contained in CM were performed using the human Aβ1-42 (Wako, Osaka, Japan) and Aβ1-40 (Immuno-Biological Lab, Gunma, Japan) assay kits according to the manufacturer’s instructions.
Reverse transcriptase-PCR and real-time PCR
Total RNA was extracted from SK-N-MC cells transduced or untransduced with lenti-APPsw-GFP or lenti-GFP, and CM-treated human NSCs under proliferating or differentiation conditions using TRI Reagent (Molecular Research Center, Cincinnati, OH, USA). RNA was quantified with a spectrophotometer, and then 1–3 μg RNA was reversely transcribed using SuperScript III Reverse Transcriptase (Invitrogen) to synthesize cDNAs following the manufacturer’s protocol. Reverse transcriptase-PCR was performed using 1 μl cDNA as follows: 95 °C for 5 min and 27 cycles of 95 °C for 30 s, 57 °C for 30 s and 72 °C for 30 s. Real-time PCR was performed using 1 μl cDNA as follows: 95 °C for 2 min and 45 cycles of 95 °C for 15 s and 60 °C for 1 min, as mentioned above. Primer sequences are shown in Supplementary Table 1. The differences in relative gene expression were normalized with glyceraldehyde 3-phosphate dehydrogenase expression and evaluated using the Advanced Relative Quantification based on the E-Method provided by Roche Applied Science.
Oligomeric Aβ42 peptides preparation
As described previously,42 synthetic human Aβ42 peptides (Invitrogen) were prepared as soluble oligomers. Briefly, the peptides were dissolved to 1 mM in 1,1,1,3,3,3-hexafluoro 2-propanol (Sigma) and lyophilized. Dried peptides were resolved to 5 mM in DMSO, diluted to 100 μM in N2 media and incubated at 4 °C for 24 h. In subsequent experiments, these oligomeric peptides were used at a final concentration of 1 μM in culture media. Control experiments were performed by adding an equal volume of DMSO used to dissolve dried Aβ42.
Statistical analysis
Statistical analyses were performed using the SPSS software (IBM Company, Armonk, NY, USA). Statistical differences were assessed with either the nonparametric Mann–Whitney U-test or the parametric unpaired t-test between two groups. Statistical analyses among at least three groups were performed using the Kruskal–Wallis test and the Mann–Whitney U-test as a post hoc test. All data are presented as means±s.e.m. Values of P<0.05 were considered to indicate significance.
Results
Aβ peptides secreted from APPsw-expressing cells exist as neurotoxic oligomers in CM
A total of 80% of SK-N-MC cells transduced with lenti-APPsw-GFP exhibited GFP-positive staining, and abundant expression of APP and Aβ (Figures 1a–e). CM derived from these APPsw-expressing cells (APP-CM) contained higher levels of Aβ40 (37.25±0.36 pg ml−1, n=3) and Aβ42 (1.32±0.01 pg ml−1) peptides than did CM from GFP-expressing cells (GFP-CM; Aβ40, 9.58±0.85 pg ml−1 and Aβ42, 0.26±0.03 pg ml−1) and SK-N-MC cells (SK-CM; Aβ40, 10.08±0.24 pg ml−1 and Aβ42, 0.25±0.01 pg ml−1; Figure 1f). Moreover, Aβ proteins secreted from APPsw-expressing cells remained as monomers and oligomers in CM (Figure 1g). APP-CM treatment significantly decreased PC12 cell survival (78.07±1.01% of SK-CM; Figure 2a). In the cortices of brain slices, APP-CM treatment markedly increased the number of TUNEL-positive cells (green) in NeuN-positive cells (red) compared with SK-CM and GFP-CM (Figures 2b–d). Arrows indicate colocalization of TUNEL- and NeuN-positive cells (Figure 2d). In contrast, the removal of Aβ in APP-CM (APP-CM-6E10) via 6E10 antibody-mediated immunoprecipitation did not elicit any neurotoxicity (Figure 2e). As a control, IgG-immunoprecipitated APP-CM (APP-CM-IgG) treatment induced colocalization of TUNEL- and NeuN-positive cells (arrows in Figure 2f). Therefore, Aβ peptides secreted from APPsw-expressing cells are naturally oligomerized in APP-CM, and these Aβ oligomers appear to be markedly neurotoxic.

Amyloid-β (Aβ) peptides secreted from APPsw-expressing cells naturally oligomerize in culture media. (a) Lenti-APPsw-GFP-, lenti-GFP-transduced and untransduced SK-N-MC cells formed spheres when cultured in N2 media to prepare conditioned media (CM). (b) Transduced cells markedly expressed GFP only. (c) Flow cytometry analysis showed that 88.5±0.3% of the APPsw-expressing cells (blue line histogram) and 92.5±0.9% of the GFP-expressing cells (green line histogram) were GFP positive. Untransduced cells were used as a negative control (black line histogram). (d) Lenti-APPsw-GFP-transduced cells transcribed abundant amyloid precursor protein (APP), and (e) highly expressed APP and Aβ proteins. (f) The quantities of Aβ40 and Aβ42 in CM were measured by enzyme-linked immunosorbent assay (ELISA). (g) Aβ peptides in CM derived from APPsw-expressing cells were identified as multiple oligomers in blots probed using the 6E10 antibody after immunoprecipitation with 6E10. Scale bar, 50 μm. APP-CM, lentiviral APPsw-GFP-transduced cell-derived CM; APPsw, amyloid precursor protein Swedish mutant; GFP, green fluorescent protein; GFP-CM, lentiviral GFP-transduced cell-derived CM; Lt APPsw-GFP, lentiviral APPsw-GFP-transduced cells; Lt GFP, lentiviral GFP-transduced cells; SK-CM, untransduced cell-derived CM; Untrans, untransduced SK-N-MC cells.

APPsw-expressing cell-derived amyloid-β (Aβ) oligomers reduce PC12 cell viability and increase the terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) positivity of cortical neurons. (a) PC12 cells co-cultured with APP-CM for 36 h had 22% reduced viability compared with cells co-cultured with SK-CM. There was no difference in the cell viability between SK-CM- and GFP-CM-treated cells. *P<0.05 compared with SK-CM- or GFP-CM-treated cells. (b–f) Representative images of brain slices exhibiting neurotoxicity from cell-derived Aβ oligomers. (b–d) In cortices of brain slices from 3-month-old B57BL6/J mice (n=8), APP-CM treatment markedly increased the number of TUNEL-positive cells (green) in NeuN-positive cells (red) compared with SK-CM and GFP-CM treatment. (e) Aβ-immunodepleted APP-CM (APP-CM-6E10) prevented apoptosis of cortical neurons. (f) In contrast, control IgG-immunoprecipitated APP-CM (APP-CM-IgG) induced colocalization of TUNEL- and NeuN-positive cells. Arrows indicate colocalization of TUNEL- and NeuN-positive cells. Scale bar, 100 μm. APP-CM, lentiviral APPsw-GFP-transduced cell-derived CM; APPsw, amyloid precursor protein Swedish mutant; CM, conditioned media; GFP, green fluorescent protein; GFP-CM, lentiviral GFP-transduced cell-derived CM; SK-CM, untransduced cell-derived CM.
CM from APPsw-expressing cells reduce proliferation and induce senescence of human NSCs
Flow cytometry analysis determined that the percentages of BrdU-positive NSCs after APP-CM and SK-CM treatments were 25.5±3.7% (n=3) and 46.5±4.3%, respectively (Figures 3a and b). In addition, the proportion of BrdU-positive NSCs between SK-CM and GFP-CM treatment had no difference (Supplementary Figure 1A). Cell cycle distribution analysis revealed a significant ∼4% increase in G0/G1-phase NSCs relative to SK-CM-treated cells after APP-CM treatment (from 79.3±0.3 to 83.7±0.7%). Concurrently, G1 arrest occurred with a considerable decrease in the proportion of G2/M and S-phase cells (from 2.4±0.2 to 0.6±0.1% and from 18.4±0.2 to 15.8±0.8%, respectively; Figure 3d). However, these CM rarely triggered death of NSCs with either APP-CM or SK-CM treatments (TUNEL+ cells<0.5% of total cells; Figure 3c). Thus, APP-CM treatment reduced the dividing potential of human NSCs via cell cycle arrest, but not via apoptosis. In addition, senescence-associated β-galactosidase activity in extracts of NSCs treated with APP-CM was markedly increased at pH 4.5 and 6.0 compared with SK-CM treatment (Figure 3e). The relative telomere length of NSCs treated with APP-CM was considerably shortened, and telomere reverse transcriptase) expression increased compared with SK-CM treatment (Figure 3f). Increased telomere reverse transcriptase levels may reflect an attempt by the cells to compensate for erosion of telomeres. Therefore, we conclude that APP-CM treatment induced senescence of NSCs via telomere dysfunction. These data demonstrate that APP-CM decreases the proliferation of human NSCs without cell death and leads to senescence through telomere shortening.

Conditioned media (CM) derived from APPsw-expressing cells reduce proliferation, induce senescence and promote telomere shortening of human neural stem cells (NSCs). (a, b) Representative histograms of flow cytometry analysis. (a) The percentages of 5′-bromo-2′-deoxyuridine (BrdU)-positive NSCs in total cells were calculated following APP-CM (green line) and SK-CM (blue line) treatments. A negative control (red line) was included without primary BrdU antibodies. (b) The percentage of BrdU-positive cells after APP-CM treatment was significantly smaller than that after SK-CM treatment. (c) In both APP-CM and SK-CM treatments, few NSCs were positive for terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL; <0.5% of total cells). (d) Cell cycle distribution of human NSCs using flow cytometry. After APP-CM treatment, NSCs in G0/G1 significantly increased by ∼4% relative to SK-CM-treated cells. Concurrently, G1 arrest occurred with a considerable decrease in the proportion of G2/M- and S-phase cells. (e) Senescence-associated β-galactosidase (SA β-gal) activity in human NSCs. APP-CM treatment significantly induced an increase in SA β-gal activity at both pH 6.0 and 4.5 compared with SK-CM treatment. (f) Relative telomere length and telomere reverse transcriptase (TERT) expression in human NSCs using real-time PCR. APP-CM treatment induced lower telomere length and higher expression of TERT in NSCs than SK-CM treatment. Mann–Whitney U-test, *P<0.05. APP-CM, lentiviral APPsw-GFP-transduced cell-derived CM; APPsw, amyloid precursor protein Swedish mutant; CM, conditioned media; GFP, green fluorescent protein; SK-CM, untransduced cell-derived CM.
CM from APPsw-expressing cells impair neurogenesis and promote gliogenesis of human NSCs
Under the differentiation conditions in culture, APP-CM- and SK-CM-treated NSCs showed the immunoreactivity for the immature cell marker nestin (32.2±1.3 vs 44.7±2.0%, respectively, n=3), early neuronal marker Tuj1 (40.7±0.9 vs 48.1±0.5%), astrocyte markers GFAP (67.5±1.6 vs 59.9±1.8%) and S100β (18.1±1.9 vs 11.5±2.2%) and oligodendrocyte precursor markers GalC (50.2±2.6 vs 31.6±2.2%) and O4 (40.1±3.5 vs 23.6±2.5%; Figures 4a–g). Intriguingly, a few nestin-positive cells coexpressed Tuj1, GFAP and GalC. Although cell fate is driven toward a neuronal or glial lineage, these colabeled cells may remain in an immature state. However, these findings indicate that APP-CM treatment induces the differentiation of NSCs into astrocytes or oligodendrocytes rather than neurons as compared with SK-CM treatment. In addition, quantitative PCR analysis determined that APP-CM treatment elicited significantly increased levels of GFAP and GalC, and decreased levels of nestin and Tuj1 in NSCs, as compared with SK-CM treatment (Figure 4h). In addition, there was no difference of fate of NSCs between the treatment of GFP-CM and SK-CM (Supplementary Figure 1B). Thus, the results suggest that APP-CM stimulates the differentiation of human NSCs, and enhances the glial cell fate, while abrogating neuronal fate of NSCs.

Conditioned media (CM) derived from APPsw-expressing cells reduce neurogenesis and promote gliogenesis of human neural stem cells (NSCs). (a–g) Representative images of differentiated NSCs in vitro. APP-CM treatment significantly altered the percentages of cells positive for nestin (red in (a)), class III β-tubulin (Tuj1; green in (b)), glial fibrillary acidic protein (GFAP; green in (c)), S100β (red in (d)), galactocerebroside (GalC; green in (e)) and O4 (red in (f)) in total 4',6-diamidino-2-phenylindole (DAPI)-positive cells (blue) compared with SK-CM treatment. APP-CM treatment induced the differentiation of NSCs into more astrocytes (c, d) and oligodendrocytes (e, f) rather than neurons (b). (h) Relative expression of cell markers in NSCs treated with APP-CM and SK-CM using real-time PCR. APP-CM treatment induced significant decreases in nestin and Tuj1, and increased levels of GFAP and GalC in NSCs compared with SK-CM treatment. Mann–Whitney U-test, *P<0.05. APP-CM, lentiviral APPsw-GFP-transduced cell-derived CM; APPsw, amyloid precursor protein Swedish mutant; CM, conditioned media; GFP, green fluorescent protein; SK-CM, untransduced cell-derived CM.
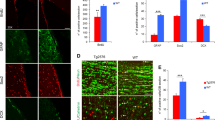
Following transplantation of human NSCs into NSE/APPsw transgenic mice brains, GFP- or BrdU-positive grafted cells migrated and engrafted into the dentate gyrus of the hippocampus at 6 weeks after grafting (Figures 5a and b). BrdU-positive cells were fully colocalized with SC121, a human-specific cytoplasm marker (Figure 5c), and gave rise to multiple neural cell lineages in both NSE/APPsw transgenic and wild-type mice brains (Figures 5d, e and g–i). In particular, counting of GFAP-positive cells among BrdU-positive cells showed a significant 10% increase in NSE/APPsw transgenic mice compared with age-matched wild-type mice (Figure 5f). Therefore, these results indicate that the Aβ-enriched milieu in NSE/APPsw transgenic mice brains may enhance astroglial differentiation of transplanted human NSCs, consistent with the in vitro results.

Enhanced astroglial differentiation of human neural stem cells (NSCs) in NSE/APPsw transgenic mice following transplantation. APPsw, amyloid precursor protein Swedish mutant; NSE, neuron-specific enolase. (a–c) Representative images of graft-derived cells. Green fluorescent protein (GFP; green in (a))- or 5′-bromo-2′-deoxyuridine (BrdU; green in (b))-positive transplanted cells showed robust migration and engraftment in the dentate gyrus of the hippocampus at 6 weeks after grafting. (c) BrdU-positive cells (green) were colocalized with human-specific cell marker SC121 (red). (d, e) Confocal microscopic images showed that some of the BrdU-positive cells (arrowheads, green) were colocalized with glial fibrillary acidic protein (GFAP)-positive cells (red) in the dentate gyri of both NSE/APPsw and age-matched wild-type mice. (f) Numbers of GFAP-positive cells among BrdU-positive grafted cells were significantly increased (by 10%) in NSE/APPsw transgenic mice (n=4) compared with age-matched wild-type mice (n=3). Mann–Whitney U-test, *P<0.05. (g–i) Transplanted human NSCs gave rise to all three neural cell types: neurons, oligodendrocytes and astrocytes in mouse brains. BrdU-positive cells (green) expressed class III β-tubulin (Tuj1; red) in wild-type mice (g) and nestin (red) in transgenic mice (i). Grafted SC121-positive cells (green) expressed olig2 (red) in transgenic mice (h). Scale bar, 100 μm in (a, b); 20 μm in (c–e, g–i).
Aβ oligomers secreted from APPsw-expressing cells modulate proliferation and differentiation of human NSCs via GSK-3β activation
APP-CM treatment significantly reduced the expression of PCNA in NSCs under the proliferation conditions. However, Aβ-immunodepleted APP-CM (APP-CM-6E10) restored the level of PCNA (Figures 6a and c). In NSCs under the differentiation conditions, the expression of PSA-NCAM, S100β and MBP induced by APP-CM treatment was also inversely regulated by Aβ-depleted APP-CM (Figures 6b and c). Thus, Aβ oligomers in APP-CM play a key role in modulating NSCs under both proliferation and differentiation conditions. To investigate the mechanism mediating the effect of Aβ oligomers in APP-CM on NSCs, NSCs were treated with both APP-CM and APP-CM containing TDZD-8 (APP-CM-TDZD-8). APP-CM treatment significantly decreased phosphorylation of GSK-3β at Ser9, an inactive form of GSK-3β (Figures 6d and e). Sequentially, activated GSK-3β reduced the level of β-catenin, a GSK-3β-targeted downstream molecule (Figures 6d and e). However, APP-CM-TDZD-8 treatment restored the levels of PCNA, PSA-NCAM, S100β and MBP, as well as GSK-3β phosphorylation and β-catenin, to that expressed after SK-CM treatment (Figures 6d and e). Thus, these data suggest that Aβ oligomers secreted from APPsw-expressing cells decrease proliferation, impair neurogenesis and promote gliogenesis of human NSCs in a GSK-3β-dependent manner. In addition, Stat3 (signal transducer and activator of transcription 3) phosphorylation of NSCs under differentiation conditions was observed after APP-CM treatment, which was inversely regulated by APP-CM-TDZD-8 treatment (Figure 6e). The phosphorylation of Stat3 is known to be involved in the gliogenesis of NSCs.43, 44

Amyloid-β (Aβ) oligomers in conditioned media (CM) derived from APPsw-expressing cells regulate the proliferation and differentiation of human neural stem cells (NSCs) via a glycogen synthase kinase-3β (GSK-3β)-mediated signaling pathway. (a, b) Western blot analysis of human NSCs. (a) In comparison with SK-CM treatment, the proliferating cell nuclear antigen (PCNA) level of NSCs under proliferative conditions was decreased by APP-CM treatment, which was restored by treatment with Aβ-neutralized APP-CM (APP-CM-6E10). IgG-immunoprecipitated APP-CM (APP-CM-mIgG) treatment was used as a control. (b) In NSCs under differentiation conditions, APP-CM treatment decreased the level of PSA-NCAM, and increased the levels of S100β and myelin basic protein (MBP) compared with SK-CM treatment. However, the levels of PSA-NCAM, S100β and MBP induced by APP-CM were inversely regulated by APP-CM-6E10. (c) Relative protein levels of PCNA, PSA-NCAM, S100β and MBP in NSCs treated with SK-CM, APP-CM and APP-CM-6E10. Mann–Whitney U-test, *P<0.05 vs SK-CM; #P<0.05 vs APP-CM; †P<0.05 vs APP-CM-6E10. (d) Under proliferative conditions, the levels of PCNA, GSK-3β phosphorylation (pGSK-3β) and β-catenin of NSCs were decreased by APP-CM treatment, and inversely regulated by treatment with APP-CM containing TDZD-8, an inhibitor of GSK-3β (APP-CM-TDZD-8). APP-CM containing DMSO (APP-CM-DMSO) was used as a control. The bars show the relative protein levels of PCNA, pGSK-3β and β-catenin (right panel). Mann–Whitney U-test, *P<0.05 vs SK-CM; #P<0.05 vs APP-CM; ††P<0.05 vs APP-CM-TDZD-8. (e) In NSCs under differentiation conditions, APP-CM treatment decreased the level of PSA-NCAM, pGSK-3β and β-catenin, and increased the levels of S100β, MBP and signal transducer and activator of transcription 3 (Stat3) phosphorylation (pStat3) compared with SK-CM treatment. The protein levels induced by APP-CM were inversely regulated by APP-CM-TDZD-8. The bars show the relative protein levels (right panel). Mann–Whitney U-test, *P<0.05 vs SK-CM; #P<0.05 vs APP-CM; ††P<0.05 vs APP-CM-TDZD-8. APP-CM, lentiviral APPsw-GFP-transduced cell-derived CM; APPsw, amyloid precursor protein Swedish mutant; DMSO, dimethyl sulfoxide; GFP, green fluorescent protein; SK-CM, untransduced cell-derived CM.
Aβ oligomers secreted from APPsw-expressing cells attenuate the migration of human NSCs via GSK-3β activation
Human NSC-derived neurospheres exposed to APP-CM and SK-CM migrated radially out of the spheres, and these cells expressed nestin, Tuj1 and GFAP (Figures 7A–D). The area of migrating cells from the neurospheres was significantly decreased after APP-CM treatment (0.14±0.01 mm2, n=10) compared with SK-CM treatment (0.23±0.02 mm2) (Figures 7Aa, Bb and E). In addition, there appeared to be no difference in the extent of cell migration from the neurospheres between GFP-CM and SK-CM treatment (Supplementary Figure 1C). Thus, APP-CM treatment inhibited the distribution of migrating cells from the edge of the neurosphere, and attenuated the migratory potential of human NSCs. In addition, when NSCs were co-cultured with APPsw-expressing cells in transwells for 3 days, the number of NSCs that migrated across the membrane was significantly decreased compared with co-culture with SK-N-MC cells on the first, second and third day, respectively (Figures 7F–H). This result suggests that Aβ released from APPsw-expressing cells may reduce the mobility of NSCs through the pores of the membrane. Furthermore, by using 6E10 antibody-mediated immunoprecipitation, the removal of Aβ peptides in APP-CM restored the migratory capacity of NSCs out of neurospheres, whereas APP-CM-IgG treatment as a control did not affect migration (Figures 7I, J and L). GSK-3β inactivation in APP-CM by TDZD-8 also increased mobility of NSCs (Figure 7K). Therefore, Aβ oligomers secreted from APPsw-expressing cells attenuated the migration of human NSCs via GSK-3β activation.

Amyloid-β (Aβ) oligomers in conditioned media (CM) derived from APPsw-expressing cells inhibit the migration of human neural stem cells (NSCs). (A–E, Aa, Bb) The migration of NSCs from neurospheres. Many of the nestin-, class III β-tubulin (Tuj1)- or glial fibrillary acidic protein (GFAP)-positive cellular processes of NSCs were migrating away from the neurosphere in SK-CM, whereas APP-CM treatment inhibited cellular migration from the neurosphere (A–D). When areas of migrating cells from neurosphere were calculated (Aa, Bb), APP-CM treatment significantly decreased cellular migrating areas compared with SK-CM (E). Unpaired t-test, *P<0.05. Scale bar, 100 μm. (F–H) The movement of NSCs across the membrane of transwells when co-cultured with APPsw-expressing and untransduced SK-N-MC cells in transwells for 3 days. Representative images of migrating 4′,6-diamidino-2-phenylindole (DAPI)-positive NSCs on the bottom of the membrane at the third day after co-culture (F, G). Compared with SK-N-MC cells, co-culture with APPsw-expressing cells significantly reduced the number of NSCs across the membrane for 3 days (214.6±21.8 vs 180.3±2.9 on the first day, 560.4±12.5 vs 291.6±37.3 on the second day and 884.3±42.4 vs 481.3±41.9 on the third day, respectively) (H). Mann–Whitney U-test, *P<0.05. Scale bar, 100 μm. (I–J) Aβ oligomers attenuated the migration of NSCs in a glycogen synthase kinase-3β (GSK-3β)-dependent manner. Aβ oligomers in CM derived from APPsw-expressing cells (APP-CM) markedly reduced cellular migration from neurosphere, whereas the removal of Aβ peptides in APP-CM by 6E10 antibody-mediated immunoprecipitation (APP-CM-6E10) restored the migratory capacity of NSCs (I, J). However, isotype-matched IgG treated APP-CM (APP-CM-IgG) as a control did not influence migration of NSCs (L). GSK-3β inactivation in APP-CM triggered by TDZD-8 increased mobility of NSCs (K). Scale bar, 50 μm. APP-CM, lentiviral APPsw-GFP-transduced cell-derived CM; APPsw, amyloid precursor protein Swedish mutant; GFP, green fluorescent protein; SK-CM, untransduced cell-derived CM.
Oligomeric forms of synthetic Aβ42 peptides modulate the proliferation, differentiation and migration patterns of human NSCs
Rather than investigating the naturally secreted Aβ oligomers from SK-N-MC cells, we examined the effects of synthetic Aβ42 peptides prepared as oligomers on the properties of NSCs. Under the same conditions described above, we evaluated the proliferation, differentiation and migration patterns of NSCs. After treatment with 1 μM of synthetic Aβ42 oligomers, NSCs under proliferative conditions showed ∼50% decrease of BrdU-positive cells relative to normal N2 media or DMSO treatment as controls (Figure 8a). Treatment with synthetic Aβ oligomers induced a significant decrease in nestin and Tuj1, and increase in GFAP and GalC, gene expression in NSCs under differentiation conditions compared with N2 media and DMSO treatment as controls (Figure 8b). Finally, the distribution of NSCs from neurospheres was markedly decreased by synthetic Aβ42 oligomers compared with treatment with N2 media and DMSO (Normal, 0.30±0.04 mm2, n=5; DMSO, 0.26±0.01 mm2; Aβ42, 0.14±0.02 mm2; Figures 8c–f). Thus, synthetic Aβ42 oligomers decrease the proliferation, promote the gliogenesis and suppress the migration of NSCs, which is consistent with the finding that Aβ oligomers in APP-CM have a marked effect on the properties of human NSCs.

Synthetic amyloid-β (Aβ) oligomers regulate the proliferation, differentiation and migration of human neural stem cells (NSCs) similar to cell-derived Aβ oligomers. (a) The number of 5′-bromo-2′-deoxyuridine (BrdU)-incorporating NSCs was reduced significantly after synthetic Aβ oligomer treatment. (b) Synthetic Aβ oligomers induced a significant decrease in nestin and class III β-tubulin (Tuj1), and an increase in glial fibrillary acidic protein (GFAP) and galactocerebroside (GalC), gene expression. (c–e) Radial migration of NSCs from neurospheres was impaired by treatment with synthetic Aβ oligomers. Scale bar, 100 μm. (f) Synthetic Aβ oligomers significantly decreased cellular migrating areas from neurospheres compared with normal N2 media or dimethyl sulfoxide (DMSO) treatment. Mann–Whitney U-test, *P<0.05.
Discussion
In this study, we examined the functional impact of cell-derived naturally secreted Aβ oligomers on the properties of NSCs isolated from human fetal telencephalon. Aβ derived from APPsw-expressing cells maintained an oligomeric as well as a monomeric state in culture media and elicited neurotoxicity, similar to previous reports.16, 45, 46 Accumulating evidence suggests that soluble Aβ oligomers strongly correlate with the cognitive decline of AD patients and may be a key toxic molecule in AD.47, 48 However, Aβ oligomers aggregated from synthetic Aβ monomers are unlikely to recapitulate the conformation of naturally aggregated Aβ oligomers through post-translational modification.21, 22 Therefore, biologically aggregated Aβ oligomers in CM derived from APPsw-expressing cells may be a reasonable model for investigation of the Aβ-related effect on NSCs. Human NSCs used in the present study were isolated from a fetal telencephalon at 13 weeks of gestation and expanded in culture as neurospheres. They were able to self-renew, differentiate into multiple neural cell types and integrate into the host neural circuitry after transplantation into the developing and adult central nervous system.49, 50 Herein, we demonstrate that Aβ oligomers derived from APPsw-expressing cells reduced the proliferation and neuronal differentiation, enhanced the glial differentiation and attenuated the migration of human NSCs in a GSK-3β-dependent manner. In addition, synthetic Aβ42 oligomers regulate the properties of human NSCs identically to cell-derived Aβ oligomers, and human NSCs grafted into the brains of APPsw transgenic mice exhibited increased astroglial differentiation.
AD brain samples and chondroitin sulfate proteoglycan NG2-positive neural progenitors from cortical tissue of AD patients exhibit high levels of GSK-3β.23, 51 Our results showed that Aβ oligomers in APP-CM activated GSK-3β in human NSCs. Activated GSK-3β stimulated the degradation of β-catenin in human NSCs under proliferative conditions. As stabilization of β-catenin in embryonic mouse brain maintains the division of cortical progenitors resulting from an increased mitotic rate,52 relatively low levels of β-catenin may inversely regulate the proliferation of human NSCs via cell cycle exit. In addition, Aβ oligomers accelerated telomere shortening and senescence, supporting reports that Aβ peptides induced senescence of endothelial and peripheral blood mononuclear cells from AD patients with excessive telomere loss,53, 54 resulting in an abrogation of cell proliferation.55 Interestingly, the death of human NSCs was negligibly induced by Aβ oligomers in this study. NSCs constitutively generate various neurotrophins against apoptosis and rarely express glutamate or voltage-gated ion channels that contribute to glutamate toxicity. Thus, Aβ oligomers may not trigger apoptosis of human NSCs because of autonomously secreted neurotrophins, unlike previous findings for neurons.45, 56
Currently, it is difficult to determine alterations in hippocampal neurogenesis in AD animal models because of differences among mouse strains, diverse APP mutant-derived Aβ contents or age variations.27, 29, 30, 35 Moreover, these differences may allow Aβ toxicity to induce neuronal dysfunction and inflammation, which could stimulate or impair neurogenesis and promote the migratory potential of NSCs toward inflamed lesions in animal brains.57, 58, 59 Under differentiation conditions, our results are consistent with previous reports that Aβ peptides impair neurogenesis of fetal telencephalon-derived human NSCs.29, 32 In contrast, Aβ42 peptides promote neurogenesis of mouse hippocampal NSCs, and the fates of rat cortical NSCs were differentially regulated by Aβ40 and 42.28, 34 These inconsistencies may result from species differences and variations among NSCs isolated from different anatomical regions. Indeed, NSCs from distinct spatiotemporal neural tissue show different behaviors, ranging from molecular mechanism to fate determination.2, 3, 60 As described above, human NSCs exposed to Aβ oligomers showed decreased β-catenin levels by activated GSK-3β under differentiation conditions. Previous studies indicated that the Wnt/β-catenin signaling pathway regulates neuronal differentiation in cortical NSCs, the embryonic cortex and the adult hippocampus.61, 62, 63 Moreover, Aβ peptides directly inhibit the Wnt/β-catenin signaling pathway through interaction with the Frizzled family of Wnt receptors in 293T cells or the induction of Dickkopf-1, a negative regulator of Wnt signaling, in cortical neurons and AD brains.24, 64 Thus, human NSCs treated with Aβ oligomers impair neurogenesis because of decreased β-catenin levels induced by activated GSK-3β, similar to NG2-positive neural progenitors from AD patients.23 Interestingly, Aβ oligomers increased the phosphorylation of Stat3, which modulates gliogenesis of NSCs via the Janus kinase/STAT signaling pathway.43, 44 Although interactions between Stat3 and GSK-3β are not directly defined in NSCs,65, 66 the inhibition of GSK-3β reduced the phosphorylated Stat3 level and gliogenesis of human NSCs cultured with Aβ oligomers in this study. In addition, the increased phosphorylation of Stat3 associated with neuronal death was recently reported in APP751SL/PS1M146L transgenic mice brains, in cortical neurons treated with Aβ and in the hippocampi of AD patients.67 However, we cannot explain how the interactions between Stat3 and GSK-3β are involved in Aβ oligomer-related gliogenesis of human NSCs in this study. Taken together, these findings provide convincing evidence that cell-derived Aβ oligomers inhibit neurogenesis of human NSCs by blocking the β-catenin signaling pathway through the activation of GSK-3β. Cell-derived Aβ oligomers also enhanced gliogenesis of human NSCs through the activation of GSK-3β and/or Stat3 signaling. Consistent with other studies that indicate that Aβ peptides impair the migration of NSCs cultured as neurospheres,29, 32 we show that cell-derived Aβ oligomers suppressed the migration of human NSCs via the GSK-3β signaling. In addition, another study reported that GSK-3β regulates the migration of radial glial cells, known as NSCs, in developing cortices.68, 69 Thus, the activation of GSK-3β by Aβ oligomers aberrantly regulates and reduces the migratory potential of human NSCs.
Transplantation of human stem cells has the potential to be a versatile therapy for various diseases. However, research on the potential therapeutic use of stem cells for AD has lagged far behind that of many other neurodegenerative disorders, because the pathogenesis of AD remains uncertain. Herein, we demonstrate that naturally secreted Aβ oligomers regulate the proliferation, differentiation and migration of human NSCs via the GSK-3β signaling pathway. These findings will contribute to the development of human NSC-based treatment for AD patients by facilitating an increased understanding of the relationships between the properties of NSCs and Aβ oligomers.
References
Breunig JJ, Haydar TF, Rakic P . Neural stem cells: historical perspective and future prospects. Neuron 2011; 70: 614–625.
Kriegstein A, Alvarez-Buylla A . The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci 2009; 32: 149–184.
Temple S . The development of neural stem cells. Nature 2001; 414: 112–117.
Lindvall O, Kokaia Z . Stem cells in human neurodegenerative disorders--time for clinical translation? J Clin Invest 2010; 120: 29–40.
Lee JP, Jeyakumar M, Gonzalez R, Takahashi H, Lee PJ, Baek RC et al. Stem cells act through multiple mechanisms to benefit mice with neurodegenerative metabolic disease. Nat Med 2007; 13: 439–447.
Tamaki SJ, Jacobs Y, Dohse M, Capela A, Cooper JD, Reitsma M et al. Neuroprotection of host cells by human central nervous system stem cells in a mouse model of infantile neuronal ceroid lipofuscinosis. Cell Stem Cell 2009; 5: 310–319.
Andres RH, Horie N, Slikker W, Keren-Gill H, Zhan K, Sun G et al. Human neural stem cells enhance structural plasticity and axonal transport in the ischaemic brain. Brain 2011; 134: 1777–1789.
Mine Y, Tatarishvili J, Oki K, Monni E, Kokaia Z, Lindvall O . Grafted human neural stem cells enhance several steps of endogenous neurogenesis and improve behavioral recovery after middle cerebral artery occlusion in rats. Neurobiol Dis 2013; 52: 191–203.
Cummings BJ, Uchida N, Tamaki SJ, Salazar DL, Hooshmand M, Summers R et al. Human neural stem cells differentiate and promote locomotor recovery in spinal cord-injured mice. Proc Natl Acad Sci USA 2005; 102: 14069–14074.
Pluchino S, Gritti A, Blezer E, Amadio S, Brambilla E, Borsellino G et al. Human neural stem cells ameliorate autoimmune encephalomyelitis in non-human primates. Ann Neurol 2009; 66: 343–354.
Holtzman DM, Morris JC, Goate AM . Alzheimer’s disease: the challenge of the second century. Sci Transl Med 2011; 3: 77sr71.
Querfurth HW, LaFerla FM . Alzheimer’s disease. N Engl J Med 2010; 362: 329–344.
Wyss-Coray T . Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med 2006; 12: 1005–1015.
Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003; 300: 486–489.
Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL et al. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J Neurosci 2004; 24: 10191–10200.
Haass C, Selkoe DJ . Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 2007; 8: 101–112.
Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002; 416: 535–539.
Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ et al. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci 2005; 8: 79–84.
Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006; 440: 352–357.
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 2008; 14: 837–842.
Reed MN, Hofmeister JJ, Jungbauer L, Welzel AT, Yu C, Sherman MA et al. Cognitive effects of cell-derived and synthetically derived Abeta oligomers. Neurobiol Aging 2011; 32: 1784–1794.
Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ . Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci USA 2011; 108: 5819–5824.
He P, Shen Y . Interruption of beta-catenin signaling reduces neurogenesis in Alzheimer’s disease. J Neurosci 2009; 29: 6545–6557.
Magdesian MH, Carvalho MM, Mendes FA, Saraiva LM, Juliano MA, Juliano L et al. Amyloid-beta binds to the extracellular cysteine-rich domain of Frizzled and inhibits Wnt/beta-catenin signaling. J Biol Chem 2008; 283: 9359–9368.
Takashima A, Honda T, Yasutake K, Michel G, Murayama O, Murayama M et al. Activation of tau protein kinase I/glycogen synthase kinase-3beta by amyloid beta peptide (25-35) enhances phosphorylation of tau in hippocampal neurons. Neurosci Res 1998; 31: 317–323.
Ly PT, Wu Y, Zou H, Wang R, Zhou W, Kinoshita A et al. Inhibition of GSK3beta-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Invest 2013; 123: 224–235.
Jin K, Galvan V, Xie L, Mao XO, Gorostiza OF, Bredesen DE et al. Enhanced neurogenesis in Alzheimer’s disease transgenic (PDGF-APPSw,Ind) mice. Proc Natl Acad Sci USA 2004; 101: 13363–13367.
Lopez-Toledano MA, Shelanski ML . Neurogenic effect of beta-amyloid peptide in the development of neural stem cells. J Neurosci 2004; 24: 5439–5444.
Haughey NJ, Nath A, Chan SL, Borchard AC, Rao MS, Mattson MP . Disruption of neurogenesis by amyloid beta-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer’s disease. J Neurochem 2002; 83: 1509–1524.
Donovan MH, Yazdani U, Norris RD, Games D, German DC, Eisch AJ . Decreased adult hippocampal neurogenesis in the PDAPP mouse model of Alzheimer’s disease. J Comp Neurol 2006; 495: 70–83.
Crews L, Adame A, Patrick C, Delaney A, Pham E, Rockenstein E et al. Increased BMP6 levels in the brains of Alzheimer’s disease patients and APP transgenic mice are accompanied by impaired neurogenesis. J Neurosci 2010; 30: 12252–12262.
Mazur-Kolecka B, Golabek A, Nowicki K, Flory M, Frackowiak J . Amyloid-beta impairs development of neuronal progenitor cells by oxidative mechanisms. Neurobiol Aging 2006; 27: 1181–1192.
Eucher JN, Uemura E, Sakaguchi DS, Greenlee MH . Amyloid-beta peptide affects viability but not differentiation of embryonic and adult rat hippocampal progenitor cells. Exp Neurol 2007; 203: 486–492.
Chen Y, Dong C . Abeta40 promotes neuronal cell fate in neural progenitor cells. Cell Death Differ 2009; 16: 386–394.
Karkkainen V, Magga J, Koistinaho J, Malm T . Brain environment and Alzheimer’s disease mutations affect the survival, migration and differentiation of neural progenitor cells. Curr Alzheimer Res 2012; 9: 1030–1042.
Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC et al. Increased hippocampal neurogenesis in Alzheimer’s disease. Proc Natl Acad Sci USA 2004; 101: 343–347.
Boekhoorn K, Joels M, Lucassen PJ . Increased proliferation reflects glial and vascular-associated changes, but not neurogenesis in the presenile Alzheimer hippocampus. Neurobiol Dis 2006; 24: 1–14.
Gil ME, Coetzer TL . Real-time quantitative PCR of telomere length. Mol Biotechnol 2004; 27: 169–172.
Criscuolo F, Bize P, Nasir L, Metcalfe NB, Foote CG, Griffiths K et al. Real-time quantitative PCR assay for measurement of avian telomeres. J Avian Biol 2009; 40: 342–347.
Hwang DY, Cho JS, Lee SH, Chae KR, Lim HJ, Min SH et al. Aberrant expressions of pathogenic phenotype in Alzheimer’s diseased transgenic mice carrying NSE-controlled APPsw. Exp Neurol 2004; 186: 20–32.
Schagger H . Tricine-SDS-PAGE. Nat Protoc 2006; 1: 16–22.
Dahlgren KN, Manelli AM, Stine Jr WB, Baker LK, Krafft GA, LaDu MJ . Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem 2002; 277: 32046–32053.
He F, Ge W, Martinowich K, Becker-Catania S, Coskun V, Zhu W et al. A positive autoregulatory loop of Jak-STAT signaling controls the onset of astrogliogenesis. Nat Neurosci 2005; 8: 616–625.
Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank DA, Rozovsky I et al. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science 1997; 278: 477–483.
Yang TT, Hsu CT, Kuo YM . Cell-derived soluble oligomers of human amyloid-beta peptides disturb cellular homeostasis and induce apoptosis in primary hippocampal neurons. J Neural Transm 2009; 116: 1561–1569.
Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB et al. Aggregation of secreted amyloid beta-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J Biol Chem 1995; 270: 9564–9570.
Lesne SE, Sherman MA, Grant M, Kuskowski M, Schneider JA, Bennett DA et al. Brain amyloid-beta oligomers in ageing and Alzheimer’s disease. Brain 2013; 136: 1383–1398.
Mc Donald JM, Savva GM, Brayne C, Welzel AT, Forster G, Shankar GM et al. The presence of sodium dodecyl sulphate-stable Abeta dimers is strongly associated with Alzheimer-type dementia. Brain 2010; 133: 1328–1341.
Kim HT, Kim IS, Lee IS, Lee JP, Snyder EY, Park KI . Human neurospheres derived from the fetal central nervous system are regionally and temporally specified but are not committed. Exp Neurol 2006; 199: 222–235.
Park S, Kim HT, Yun S, Kim IS, Lee J, Lee IS et al. Growth factor-expressing human neural progenitor cell grafts protect motor neurons but do not ameliorate motor performance and survival in ALS mice. Exp Mol Med 2009; 41: 487–500.
Lee HK, Kumar P, Fu Q, Rosen KM, Querfurth HW . The insulin/Akt signaling pathway is targeted by intracellular beta-amyloid. Mol Biol Cell 2009; 20: 1533–1544.
Chenn A, Walsh CA . Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 2002; 297: 365–369.
Donnini S, Solito R, Cetti E, Corti F, Giachetti A, Carra S et al. Abeta peptides accelerate the senescence of endothelial cells in vitro and in vivo, impairing angiogenesis. FASEB J 2010; 24: 2385–2395.
Damjanovic AK, Yang Y, Glaser R, Kiecolt-Glaser JK, Nguyen H, Laskowski B et al. Accelerated telomere erosion is associated with a declining immune function of caregivers of Alzheimer’s disease patients. J Immunol 2007; 179: 4249–4254.
Ross HH, Levkoff LH, Marshall GP 2nd, Caldeira M, Steindler DA, Reynolds BA et al. Bromodeoxyuridine induces senescence in neural stem and progenitor cells. Stem Cells 2008; 26: 3218–3227.
Mattson MP . Pathways towards and away from Alzheimer’s disease. Nature 2004; 430: 631–639.
Nakatomi H, Kuriu T, Okabe S, Yamamoto S, Hatano O, Kawahara N et al. Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell 2002; 110: 429–441.
Monje ML, Toda H, Palmer TD . Inflammatory blockade restores adult hippocampal neurogenesis. Science 2003; 302: 1760–1765.
Imitola J, Raddassi K, Park KI, Mueller FJ, Nieto M, Teng YD et al. Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1alpha/CXC chemokine receptor 4 pathway. Proc Natl Acad Sci USA 2004; 101: 18117–18122.
Ming GL, Song H . Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 2011; 70: 687–702.
Lie DC, Colamarino SA, Song HJ, Desire L, Mira H, Consiglio A et al. Wnt signalling regulates adult hippocampal neurogenesis. Nature 2005; 437: 1370–1375.
Hirabayashi Y, Itoh Y, Tabata H, Nakajima K, Akiyama T, Masuyama N et al. The Wnt/beta-catenin pathway directs neuronal differentiation of cortical neural precursor cells. Development 2004; 131: 2791–2801.
Munji RN, Choe Y, Li G, Siegenthaler JA, Pleasure SJ . Wnt signaling regulates neuronal differentiation of cortical intermediate progenitors. J Neurosci 2011; 31: 1676–1687.
Caricasole A, Copani A, Caraci F, Aronica E, Rozemuller AJ, Caruso A et al. Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is associated with neuronal degeneration in Alzheimer’s brain. J Neurosci 2004; 24: 6021–6027.
Beurel E, Jope RS . Differential regulation of STAT family members by glycogen synthase kinase-3. J Biol Chem 2008; 283: 21934–21944.
Zhu Z, Kremer P, Tadmori I, Ren Y, Sun D, He X et al. Lithium suppresses astrogliogenesis by neural stem and progenitor cells by inhibiting STAT3 pathway independently of glycogen synthase kinase 3 beta. PLoS One 2011; 6: e23341.
Wan J, Fu AK, Ip FC, Ng HK, Hugon J, Page G et al. Tyk2/STAT3 signaling mediates beta-amyloid-induced neuronal cell death: implications in Alzheimer’s disease. J Neurosci 2010; 30: 6873–6881.
Yokota Y, Eom TY, Stanco A, Kim WY, Rao S, Snider WD et al. Cdc42 and Gsk3 modulate the dynamics of radial glial growth, inter-radial glial interactions and polarity in the developing cerebral cortex. Development 2010; 137: 4101–4110.
Asada N, Sanada K . LKB1-mediated spatial control of GSK3beta and adenomatous polyposis coli contributes to centrosomal forward movement and neuronal migration in the developing neocortex. J Neurosci 2010; 30: 8852–8865.
Acknowledgements
This work was supported by grants from the Korea Healthcare Technology R&D Project (A091159, A121943), Ministry for Health and Welfare, the National Research Foundation of Korea (2010-0020289), Ministry of Science and Technology and the Faculty Grant of Yonsei University College of Medicine (6-2006-0017), Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Experimental & Molecular Medicine website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Lee, IS., Jung, K., Kim, IS. et al. Amyloid-β oligomers regulate the properties of human neural stem cells through GSK-3β signaling. Exp Mol Med 45, e60 (2013). https://doi.org/10.1038/emm.2013.125
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/emm.2013.125
Keywords
This article is cited by
-
The interaction between ageing and Alzheimer's disease: insights from the hallmarks of ageing
Translational Neurodegeneration (2024)
-
Stem Cells and the Microenvironment: Reciprocity with Asymmetry in Regenerative Medicine
Acta Biotheoretica (2022)
-
Amyloid β Peptide Compromises Neural Stem Cell Fate by Irreversibly Disturbing Mitochondrial Oxidative State and Blocking Mitochondrial Biogenesis and Dynamics
Molecular Neurobiology (2019)
-
Aβ42 Peptide Promotes Proliferation and Gliogenesis in Human Neural Stem Cells
Molecular Neurobiology (2019)
-
Role of Amyloid Precursor Protein (APP) and Its Derivatives in the Biology and Cell Fate Specification of Neural Stem Cells
Molecular Neurobiology (2018)

{kind=link}