
Abstract
To date, more than 30 antibodies have been approved worldwide for therapeutic use. While the monoclonal antibody market is rapidly growing, the clinical use of therapeutic antibodies is mostly limited to treatment of cancers and immunological disorders. Moreover, antibodies against only five targets (TNF-α, HER2, CD20, EGFR, and VEGF) account for more than 80 percent of the worldwide market of therapeutic antibodies. The shortage of novel, clinically proven targets has resulted in the development of many distinct therapeutic antibodies against a small number of proven targets, based on the premise that different antibody molecules against the same target antigen have distinct biological and clinical effects from one another. For example, four antibodies against TNF-α have been approved by the FDA -- infliximab, adalimumab, golimumab, and certolizumab pegol -- with many more in clinical and preclinical development. The situation is similar for HER2, CD20, EGFR, and VEGF, each having one or more approved antibodies and many more under development. This review discusses the different binding characteristics, mechanisms of action, and biological and clinical activities of multiple monoclonal antibodies against TNF-α, HER-2, CD20, and EGFR and provides insights into the development of therapeutic antibodies.
Similar content being viewed by others

Introduction
The therapeutic potential of monoclonal antibodies had been well recognized by the pharmaceutical industry, and just one decade after the development of hybridoma technology by Milstein and Köhler (Köhler and Milstein, 1975), the first therapeutic monoclonal antibody (muromonab, Orthoclone OKT3) was approved for clinical use in 1986. Subsequent technological advances such as chimerization/humanization of murine antibodies, transgenic mice, and antibody phage display (Clark, 2000) have enabled the discovery, engineering, and development of monoclonal antibodies with high efficacy and low side effects, especially in terms of immunogenicity. Recent advancements in this area include antibody-drug conjugates (ADCs) (Carter and Senter, 2008), bispecific antibodies (Müller and Kontermann, 2010), and Fc engineering for longer half-life and greater effector functions (Kaneko and Niwa, 2011). Using currently available technological platforms, it is now possible to produce highly functional antibodies against virtually any antigen or epitope. However, until recently, the number of clinically successful target antigens to which these technologies can be applied was surprisingly small. As a result, only a handful of therapeutically relevant antigens, including cell-surface proteins HER2, CD20 and EGFR, and soluble ligands TNF-α and VEGF, have been targeted by multiple antibodies, with great clinical and commercial success. While these antibodies target the same antigen, their biological and clinical characteristics, as well as their modes of action in many cases, differ widely from one another, hence justifying attempts to develop new candidate antibodies against antigens that have already been targeted by other approved antibody drugs. Detailed comparisons of antibodies that target the same antigen (TNF-α, HER2, EGFR or CD20) are given in this review, with emphases on their biochemical/biophysical properties and mechanisms of action (Figure 1).

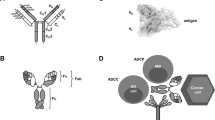
Mechanisms of action for therapeutic antibodies. Antibodies against soluble ligands, such as anti-TNF-α antibodies infliximab, adalimumab, golimumab and certolizumab pegol, interfere with ligand-receptor interaction (A). Anti-EGFR antibodies cetuximab, panitumumab and nimotuzumab inhibit ligand binding to the receptor (A) and thus stabilize the inactive conformation of EGFR (B). HER2 is in a constitutively active conformation, and anti-HER2 antibodies trastuzumab and pertuzumab block homo- and heterodimerization of HER2 with ErbB recetors (C). For antibodies targeting CD20, which does not have a known ligand and probably is not a receptor, the major mechanisms of action is Fc-mediated effector functions (D). Most of other antibodies, especially of IgG1 subtype, that bind a cell surface antigen can also mediate ADCC/CDC for effective cell killing. See text for various other possible mechanisms not shown in this figure, such as receptor internalization and sensitization of the target cells.
TNF-α
TNF-α is the single most successful antibody target molecule, worth more than 15 billion USD in combined worldwide sales in 2010 alone. There are three anti-TNF-α IgG1 antibodies (infliximab/Remicade, adalimumab/Humira, and golimumab/Simponi), one pegylated antibody fragment (certolizumab pegol/Cimzia), and an antibody-like Fc-fusion protein (etanercept/Enbrel) approved for the treatment of various autoimmune disorders. The approved indications for these molecules include rheumatoid arthritis, psoriasis, psoriatic arthritis, Crohn's disease, ulcerative colitis, and ankylosing spondylitis (Williams et al., 2007). TNF-α is expressed as a homotrimeric transmembrane protein on activated macrophages and T lymphocytes. Proteolytic cleavage of the extracellular domain releases soluble trimeric TNF-α, and both membranous and soluble TNFs are able to bind TNF receptors (TNFR1 and TNFR2). Upon binding to TNFR, TNF-α mediates apoptosis and inflammation and regulates immune functions by activating NF-κB, the MAPK pathways, and death signaling. As a master pro-inflammatory cytokine, TNF-α plays a protective role against infection and injury in normal immune responses; however, chronically elevated levels of TNF-α have also been associated with the pathogenesis of many autoimmune and inflammatory diseases (Feldmann et al., 1996; Ritchlin et al., 1998). While there are many TNF-α antagonistic antibodies, their modes of action are basically the same, i.e., inhibition of the TNF-TNFR interaction. The efficacies of these agents, therefore, are mostly determined by factors other than their modes of action, such as affinity, immunogenicity, tissue penetration, and serum half-life. While there is no head-to-head, direct comparison clinical study featuring anti-TNF-α agents, several meta-analyses have suggested that their efficacies for rheumatoid arthritis are similar to one another (Alonso-Ruiz et al., 2008; Kristensen et al., 2007; Launois et al., 2011).
It is difficult to directly compare the immunogenicity data of different antibodies from different studies since the patient groups, assays used, and criteria for determining immunogenicity vary among the studies (Emi Aikawa et al., 2010). Given this limitation in interpreting the immunogenicity data, it is generally accepted that infliximab, a mouse-human chimeric antibody with human constant regions (~75% of the immunoglobulin sequence) and mouse variable regions (~25% of the sequence), is more immunogenic than humanized or human antibody agents such as adalimumab, golimumab, and certolizumab pegol (Yoon et al., 2010). The incidence of human anti-chimeric antibody (HACA) reaction by infliximab ranges from 3% to 53% depending on the dosage and drug combination, which correlates with a reduced duration of response to treatment (Maini et al., 1998). Fully human anti-TNF-α antibodies, adalimumab and golimumab, show markedly lower immunogenicity profiles, with a 5-12% HAHA (human anti-human antibody) response for adalimumab (Abbott, 2002) and 4% for golimumab (Centocor, 2009). Similarly, 8% of patients developed immunogenicity upon treatment with certolizumab pegol, a humanized antibody (Rivkin, 2009; UCB, 2008).
The affinity of these antibodies is in the picomolar range, although some variations exist in the reported values depending on the assay format; Kd values of these antibodies against soluble TNF-α are 127 pM, 44 pM, 18 pM, and 90 pM for adalimumab, infliximab, golimumab (Shealy et al., 2010), and certolizumab pegol (UCB, 2008), respectively. While the affinities of these antibodies are similar, there are some differences in their antigen-binding profiles (Kim et al., 2007; Kohno et al., 2007). Infliximab binds trimeric soluble TNF-α at a 2:1 ratio in vitro, with a complex of six antibody molecules and three trimeric TNF-α being the dominant species. Adalimumab also is known to form high molecular weight complexes with the trimeric antigen. It is likely that golimumab also has a similar binding stoichiometry, as these IgG1 molecules are bivalent. As a result, various combinations of antibody: trimeric antigen interactions are possible. On the other hand, certolizumab pegol is a monovalent, pegylated Fab' and thus does not cross-link antigens to form large supramolecular complexes. Together with its lack of Fc-mediated effector function, this binding characteristic of certolizumab pegol may explain some of its distinct biological effects such as better tissue penetration, lack of ADCC and CDC, and reduced neutrophil degranulation and superoxide production (Nesbitt et al., 2007; Cassinotti et al., 2008; Palframan et al., 2009; Launois et al., 2011).
HER2
HER2 overexpression is found in ~30% of human breast cancers and is associated with poor disease prognosis (Hudis, 2007). Two therapeutic antibodies targeting HER2 are discussed below: trastuzumab (Herceptin) and pertuzumab (Omnitarg). Unlike TNF-α inhibitors, which function via the essentially same mechanism, these two antibodies bind to distinct epitopes on HER2 and have different mechanisms of action. Receptor tyrosine kinases (RTKs) such as HER2 and EGFR have a relatively large extracellular domain (ECD) consisting of multiple sub-domains, and they undergo multistep activation processes that include ligand binding, conformational changes, and homo/heterodimerization (Schmitz and Ferguson, 2009). Antibody binding to different parts of RTKs can thus affect receptor function and signaling in different ways.
Despite being one of the most successful monoclonal antibody therapies, the mechanism of action for trastuzumab has not yet been fully elucidated. Trastuzumab is a humanized IgG1 antibody that binds HER2 with a Kd of ~5 nM (Genentech, 2002), which is considered moderate compared to many other therapeutic antibodies with a sub-nanomolar Kd. Indeed, it has been reported that anti-HER2 antibodies with moderate affinity are characterized by greater tumor accumulation and tissue penetration, whereas high-affinity antibodies tend to localize in the perivascular region of a tumor and do not penetrate well into the tumor interior (Rudnick et al., 2011). This is also in contrast with antibodies targeting soluble ligands, such as anti-TNF-α agents, which typically have sub-nanomolar affinity and can bind and neutralize ligands at low serum concentration. While it is known that trastuzumab binds to domain IV of HER2-ECD, the exact molecular consequence of trastuzumab binding to HER2 is still under debate. Recent studies reported that trastuzumab inhibited HER2 homodimer-mediated cell growth (Ghosh et al., 2011) as well as ligand-independent HER2-HER3 heterodimerization (Junttila et al., 2009). Antibody-dependent cellular cytotoxicity (ADCC) is thought to mediate the effects of trastuzumab (Lewis et al., 1993; Cooley et al., 1999; Clynes et al., 2000; Gennari et al., 2004; Arnould et al., 2006; Barok et al., 2007; Valabrega et al., 2007). However, the relative importance of ADCC in trastuzumab action is debatable since ~70% of patients with HER2 overexpressing metastatic breast cancer did not respond to trastuzumab monotherapy (Vogel et al., 2002). Interpreting the effects of ADCC is further complicated by the fact that many cancer patients have impaired immune function due to the advanced stage of the disease or previous cancer therapies (Valabrega et al., 2007). A recent study suggested that the varying responses of patients with HER2-positive cancer to trastuzumab may be partly due to a difference in FcγRIIIa genotype, suggesting an important role for ADCC in the clinical effects of trastuzumab (Musolino et al., 2008). Downregulation of HER2 via internalization and degradation by trastuzumab and consequent reduction in receptor dimerization and signaling has also been reported as a possible mechanism of action of trastuzumab (zum Buschenfelde et al., 2002). Trastuzumab blocks shedding of HER2, which is thought to be related to its antitumor activity (Molina et al., 2001), and inhibits angiogenesis (Izumi et al., 2002; Klos et al., 2003). Another proposed mechanism for trastuzumab is inhibition of the PI3K/Akt pathway (Nagata et al., 2004). Trastuzumab binding to HER2 inhibits binding of Src to the cytoplasmic domain of HER2, thus reducing Src activity, which in turn reduces the tyrosine phosphorylation of PTEN. Dephosphorylated PTEN is subsequently recruited to the plasma membrane and activated, which in turn dephosphorylates and inactivates Akt, leading to apoptosis and inhibition of proliferation. Finally, inhibition of Akt by trastuzumab allows p27kip1 to enter the nucleus and inhibit cdk2, resulting in cell cycle arrest at G1 phase (Kute et al., 2004).
Another humanized anti-HER2 antibody, pertuzumab, binds to an epitope in domain II of HER2-ECD with a Kd of ~2.2 nM and inhibits the ligand-dependent heterodimerization of HER2 with other ErbB receptors such as EGFR and HER3 (Persson et al., 2005). HER2 is the preferred dimerization partner of HER3, which has impaired tyrosine kinase activity, and the PI3K/Akt pathway activated by the HER2/HER3 heterodimer is one of the most important HER2-related cancer cell survival/proliferation mechanisms (Hynes and MacDonald, 2009). Structural analysis of pertuzumab-HER2 binding has shown that the antibody sterically interferes with receptor dimerization and subsequent signaling (Franklin et al., 2004). By blocking HER2 heterodimerization, pertuzumab shows biological effects against cancers that do not overexpress HER2 (Agus et al., 2002). Unlike trastuzumab, which is substantially less effective when tested as the F(ab')2 fragment (Spiridon et al., 2004) or in mice lacking Fc receptor (Clynes et al., 2000), the Fab fragment of pertuzumab is as effective as the IgG1 form in inhibiting the growth of HER2 + tumors (Agus et al., 2002), implying that pertuzumab may be less dependent on ADCC than trastuzumab. On the other hand, pertuzumab does not inhibit the ligand-independent activation of HER2 as trastuzumab does. With different epitopes and mechanisms of action, the two antibodies synergistically inhibit the survival of BT474 human breast cancer cells in vitro (Nahta et al., 2004).
The different mechanisms of action and clinical effects of the two anti-HER2 antibodies suggests the possibility of developing antibodies against novel HER2 neutralizing epitopes that function differently from trastuzumab and pertuzumab. Recently, bacterial cell display-based screening of HER2 peptides revealed several new epitopes that can be targeted by antibodies to inhibit cell growth and proliferation (Rockberg et al., 2008, 2009). While it remains unclear whether or not antibodies against those epitopes really are different from trastuzumab or pertuzumab in terms of mechanism of action, the multistep activation mechanisms of RTKs such as HER2 and EGFR can be exploited to develop neutralizing antibodies with unique biochemical and clinical properties.
EGFR
Epidermal growth factor receptor (EGFR) is overexpressed in various cancers, including colorectal cancer, head-and-neck cancer, and non-small cell lung carcinoma (Nicholson et al., 2001). Although EGFR is expressed in many normal tissues, targeting of EGFR with neutralizing antibodies has proven to be effective in extending the survival of certain cancer patients (Cunningham et al., 2004; Cohenuram and Saif, 2007). Two anti-EGFR antibodies, cetuximab (Erbitux) and panitumumab (Vectibix), have been approved for the treatment of colorectal and head-and-neck cancers. Both of them bind to the same domain (domain III of EGFR ECD) and inhibit receptor activation and signaling (Li et al., 2005; Freeman et al., 2008). Nimotuzumab is another anti-EGFR antibody that has been approved in Europe and several other countries and is in phase II trials in the U.S. Several other anti-EGFR antibodies such as zalutumumab and necitumumab are also in various stages of development.
Cetuximab is a mouse-human chimeric IgG1 with high affinity (Kd ~200 pM) (Goldstein et al., 1995) and has been approved for the treatment of metastatic colorectal cancer (mCRC) and squamous cell carcinoma of the head and neck (SCCHN) as a monotherapy or in combination with irinotecan (for mCRC) or radiotherapy (for SCCHN). Upon binding to EGFR, cetuximab directly blocks ligand binding and locks the receptor in an autoinhibitory conformation, thereby preventing the receptor from assuming the extended conformation required for dimerization (Li et al., 2005). As a result, downstream signaling pathways such as the JAK/STAT, PI3K/Akt, and MAPK pathways are blocked, which inhibits cancer cell survival and proliferation (Baselga, 2001; Meira et al., 2009). Cetuximab also causes cell cycle arrest similarly to trastuzumab by increasing the expression of p27kip1, which then inhibits cdk2 in the nucleus (Kiyota et al., 2002; Wu et al., 1996). It also reduces angiogenesis (Perrotte et al., 1999; Petit et al., 1997) and metastasis (Huang et al., 2002) by decreasing the levels of angiogenic factors and matrix metalloproteinases. Antibody-dependent cellular cytotoxicity (Kawaguchi et al., 2007; Kurai et al., 2007), induction of apoptosis by increased expression of the proapoptotic protein Bax (Mandal et al., 1998), and sensitization of cancer cells to chemotherapeutic agents (Mahtani and Macdonald, 2008) or radiation (Gebbia et al., 2007) are other mechanisms of action that have been proposed for cetuximab.
Panitumumab is a fully human IgG2 antibody made by XenoMouse transgenic technology (Jakobovits et al., 2007). It binds to domain III of EGFR with an affinity higher than that of cetuximab (Kd ~50 pM) (Kim and Grothey, 2008), and it functions by blocking ligand binding to EGFR (Freeman et al., 2008; Schmitz and Ferguson, 2009). Similar to cetuximab, the suggested mechanisms of action for panitumumab include decreased EGFR signaling, cell cycle arrest, reduced angiogenesis, and receptor downregulation by internalization (Messersmith and Hidalgo, 2007; Yang et al., 1999). As it has a fully human immunoglobulin sequence, panitumumab has lower immunogenicity compared to cetuximab (Yoon et al., 2010). Since it is an IgG2, panitumumab is also expected to have much weaker effector functions than cetuximab and be less likely to damage normal EGFR-expressing cells, although a recent study indicated that panitumumab is effective in triggering ADCC by neutrophils and monocytes (Schneider-Merck et al., 2010).
Nimotuzumab is a humanized IgG1 antibody with a much lower affinity than both cetuximab and panitumumab. Probably as a result of this, it shows antitumor activity without the severe skin rashes commonly associated with other anti-EGFR antibodies (Ramakrishnan et al., 2009). Due to its lower affinity, nimotuzumab can form stable bivalent attachments to EGFR-overexpressing cells, but not to cells with a normal level of EGFR expression, minimizing skin toxicity. The low-affinity strategy has also been employed in the development of adecatumumab, which targets a pan-epithelial antigen EpCAM. These antibodies exemplify the fine balance between affinity, toxicity, and distribution required when developing therapeutic antibodies targeting a cell surface antigen.
For anti-EGFR antibodies, KRAS mutation is a predictive biomarker of resistance to antibody treatment (Heinemann et al., 2009; Lièvre et al., 2006). KRAS is a small G protein that is critically involved in the Ras/MAPK pathway downstream of EGFR. Mutation of KRAS activates this signaling pathway even in the presence of EGFR-neutralizing antibodies. On the other hand, the EGFR expression level (as determined by IHC) is not correlated with the response to anti-EGFR antibody therapy (Cunningham et al., 2004; Saltz et al., 2004), and some patients with EGFR-negative tumors have been reported to respond to anti-EGFR therapy (Chung et al., 2005). The reason for this apparent paradox is not yet clear, although it is generally undisputed that cetuximab and panitumumab exert clinical efficacy exclusively by binding to EGFR. Whatever the reason, unlike KRAS mutation status, the EGFR expression level is not considered to be a predictive biomarker when cancer patients are subjected to anti-EGFR antibody therapy. Novel or alternative therapeutic options are being investigated in patients with KRAS mutation for whom cetuximab or panitumumab treatment offers little or no clinical benefit (Dunn et al., 2011; Lee et al., 2011).
CD20
CD20 is a putative tetra-transmembrane cell surface antigen expressed only on B cell precursors and mature B cells. Its function has not yet been clearly elucidated, but studies have suggested that CD20 is involved in B cell growth and differentiation and probably functions as a calcium channel (Beers et al., 2010). The selective depletion of CD20+ cells does not result in excessive toxicity (McLaughlin et al., 1998) since pre-existing plasma cells as well as hematopoietic stem cells lack CD20, and also normal B cells are replenished by stem cell differentiation. This makes CD20 an ideal target for treatment of cancers of B cell lineage such as non-Hodgkin's lymphoma (NHL) and chronic lymphocytic leukemia (CLL). Currently, there are four approved antibody drugs targeting CD20. Ibritumomab tiuxetan (Zevalin) and tositumomab-I131 (Bexxar) are murine antibody radioimmunoconjugates. Rituximab (Rituxan/MabThera) is a human-mouse chimeric IgG1 antibody approved for the treatment of NHL, CLL, and rheumatoid arthritis (RA), as well as Wegener's granulomatosis and microscopic polyangiitis. Ofatumumab (Arzerra) is a fully human IgG1 monoclonal antibody for CLL. While all of these target and deplete CD20+ cells, they differ from one another in preclinical and clinical characteristics.
Rituximab is the first antibody drug approved for an oncological indication. It binds to the large extracellular loop of CD20 (Teeling et al., 2006) with a binding affinity of approximately ~8 nM (Genentech, 1997). ADCC is thought to be the major mechanism of action for rituximab (Clynes et al., 2000; Flieger et al., 2000). CD20 is neither shed nor internalized upon antibody binding (Einfeld et al., 1988; Press et al., 1987), allowing effective cell killing by ADCC. Being a type I antibody that induces CD20 redistribution into lipid rafts (Cragg and Glennie, 2004), rituximab also strongly elicits complement-dependent cytotoxicity (CDC) by clustering multiple Fc regions (Cragg et al., 2003). This antibody is also reported to induce apoptosis of CD20+ cells (Byrd et al., 2002; Pedersen et al., 2002), although the nature and significance of this mode of action remain controversial. Finally, rituximab sensitizes cancerous cells to radio- or chemotherapy (Demidem et al., 1997; Skvortsova et al., 2006) and is used not only as a single agent but also in combination with chemotherapeutic agents.
Ofatumumab is another type I anti-CD20 monoclonal antibody. It is a fully human antibody made from a transgenic mouse expressing human antibodies. Its epitope encompasses both the small and large extracellular loops of CD20 (Uchiyama et al., 2010). With a binding affinity of ~5 nM, ofatumumab also has a slower off-rate compared to rituximab (Teeling et al., 2004). Its distinct epitope and slow dissociation allow the antibody to bind much closer as well as longer to the cell surface, which may explain more rapid complement induced cell killing by ofatumumab than rituximab (Beum et al., 2008). In fact, when tested on CEM cells transduced with human CD20, ofatumumab induced CDC better than rituximab, showing lytic activity against cells expressing as few as 4,500 CD20 molecules per cell and reaching full activity at a CD20 density of 60,000 molecules/cell. On the other hand, rituximab requires at least 30,000 CD20 molecules per cell to show lytic activity and does not achieve full lysis even at 135,000 CD20 molecules/cell (Teeling et al., 2006). Studies suggest that ofatumumab also more potently induces ADCC than rituximab by binding more tightly to FcγRIIIa on NK cells (Craigen et al., 2009).
Unlike rituximab and ofatumumab, tositumomab is a type II antibody that does not induce CD20 redistribution into lipid rafts (Beers et al., 2010). Type II antibodies are not strong activators of CDC but instead induce a unique mode of cell death involving homotypic adhesion of B cells. The existence of multiple functional epitopes that drive different cellular responses upon antibody binding, on an antigen with relatively small extracellular domains, illustrates the point that antibodies with novel mechanisms of action and improved efficacies can be developed against an established target antigen.
Antibodies in development: Future directions
Three therapeutic antibodies, each against a different new target, were approved in 2004: cetuximab (Erbitux) targeting EGFR, bevacizumab (Avastin) targeting VEGF, and natalizumab (Tysabri) targeting VLA-4. Each of these antibodies recorded >1 billion USD in global sales in 2009. No new antigen with blockbuster potential was targeted by an approved antibody until 2010, when denosumab (Prolia/Xgeva) targeting RANKL and tocilizumab (Actemra) targeting IL-6 were approved. These were soon followed in 2011 by belimumab (Benlysta) against BLyS and ipilimumab (Yervoy) against CTLA-4. It is likely that the therapeutic potential of more new targets, such as beta amyloid and VEGFR-2, will be realized in coming years. On the other hand, efforts are continuing to develop antibodies against existing targets. Particularly, many antibodies are under development against four of the most successful antibody targets (TNF-α, HER2, EGFR, and CD20) discussed in this review, reflecting the clinical validity and commercial value of these molecules. Antibodies and antibody fragments/derivatives in clinical development targeting these four antigens are summarized in Table 1. For cell-surface antigens (HER2, EGFR, and CD20), there is an emphasis on enhancing the biological effects of antibodies by strategies such as Fc modification (including glycoengineering), antibody-drug conjugation, and simultaneous targeting of more than one target. This trend reflects the accepted view that for these well-proven oncology targets, enhanced cell killing ability of the drugs is positively correlated with therapeutic efficacy. In contrast, for the soluble ligand TNF-α, the trend is apparently focused on the development of antibody fragments and domain antibodies that are significantly smaller than immunoglobulins. With essentially identical mechanisms of action and no requirement for cytolytic or cytostatic activities, new anti-TNF-α molecules must gain a competitive advantage over existing antibodies in areas such as affinity, serum half-life, immunogenicity, tissue distribution, and development/production cost. Certolizumab pegol (a pegylated Fab'), for example, has a unique distribution profile (Palframan et al., 2009) and can be produced less expensively from a bacterial host. Therefore, along with first-in-class antibodies, more therapeutic antibodies that target established antigens with improved and/or unique therapeutic profiles are expected to enter the market in the near future.
Concluding remarks
Multiple therapeutic antibodies against one soluble ligand (TNF-α) and three cell surface proteins (HER2, EGFR, and CD20) were discussed in this review. While TNF-α is targeted by as many as four antibodies plus etanercept (a soluble receptor-Fc fusion protein), their mechanisms of action are essentially the same, i.e., blockade of ligand binding to the cell surface receptor. In contrast, antibodies against a cell surface antigen tend to have unique mechanisms of action or different in vitro and in vivo characteristics, as evidenced by trastuzumab/pertuzumab and rituximab/ofatumumab. This may be explained by the fact that an antibody can influence cell survival and proliferation in many different ways by directly binding to the cell surface. While targeting a soluble ligand may sometimes be advantageous in such aspects as toxicity, specificity, and delivery, targeting a membrane receptor is much more likely to yield antibodies that are mechanistically and clinically distinct from existing molecules. It is thus expected that scientific and technological advancements in the field will continue to allow generation of novel agents that can overcome or supplement the limitations of existing drugs.
Abbreviations
- ADC:
-
antibody-drug conjugate
- ADCC:
-
antibody-dependent cellular cytotoxicity
- CD20:
-
cluster of differentiation 20
- CDC:
-
complement dependent cytotoxicity
- CLL:
-
chronic lymphocytic leukemia
- ECD:
-
extracellular domain
- EGFR:
-
epidermal growth factor receptor
- EpCAM:
-
epithelial cell adhesion molecule
- FcγR:
-
Fc gamma receptor
- FDA:
-
Food and Drug Administration
- HACA:
-
human anti-chimeric antibody
- HAHA:
-
human anti-human antibody
- HER2:
-
human epidermal growth factor receptor 2
- IHC:
-
immunohistochemistry
- JAK:
-
Janus kinase
- KRAS:
-
V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog
- MAPK:
-
mitogen-activated protein kinase
- mCRC:
-
metastatic colorectal cancer
- NHL:
-
non-Hodgkin's lymphoma
- PI3K:
-
phosphoinositide 3-kinase
- PTEN:
-
phosphatase and tensin homolog
- RA:
-
rheumatoid arthritis
- RTK:
-
receptor tyrosine kinase
- SCCHN:
-
squamous cell carcinoma of the head and neck
- STAT:
-
signal transducer and activator of transcription
- TNFR:
-
tumor necrosis factor receptor
- VEGF:
-
vascular endothelial growth factor
References
Abbott . Humira full prescribing information . 2002, http://www.rxabbott.com/pdf/humira.pdf
Agus DB, Akita RW, Fox WD, Lewis GD, Higgins B, Pisacane PI, Lofgren JA, Tindell C, Evans DP, Maiese K, Scher HI, Sliwkowski MX . Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth . Cancer Cell 2002 ; 2 : 127 - 137
Alonso-Ruiz A, Pijoan JI, Ansuategui E, Urkaregi A, Calabozo M, Quintana A . Tumor necrosis factor alpha drugs in rheumatoid arthritis: systematic review and metaanalysis of efficacy and safety . BMC Musculoskelet Disord 2008 ; 9 : 52
Arnould L, Gelly M, Penault-Llorca F, Benoit L, Bonnetain F, Migeon C, Cabaret V, Fermeaux V, Bertheau P, Garnier J, Jeannin JF, Coudert B . Trastuzumab-based treatment of HER2-positive breast cancer: an antibody-dependent cellular cytotoxicity mechanism ? Br J Cancer 2006 ; 94 : 259 - 267
Barok M, Isola J, Palyi-Krekk Z, Nagy P, Juhasz I, Vereb G, Kauraniemi P, Kapanen A, Tanner M, Szollosi J . Trastuzumab causes antibody-dependent cellular cytotoxicity-mediated growth inhibition of submacroscopic JIMT-1 breast cancer xenografts despite intrinsic drug resistance . Mol Cancer Ther 2007 ; 6 : 2065 - 2072
Baselga J . The EGFR as a target for anticancer therapy - focus on cetuximab . Eur J Cancer 2001 ; 37 : S16 - S22
Beers SA, Chan CH, French RR, Cragg MS, Glennie MJ . CD20 as a target for therapeutic type I and II monoclonal antibodies . Semin Hematol 2010 ; 47 : 107 - 114
Beum PV, Lindorfer MA, Beurskens F, Stukenberg PT, Lokhorst HM, Pawluczkowycz AW, Parren PW, van de, Taylor RP . Complement activation on B lymphocytes opsonized with rituximab or ofatumumab produces substantial changes in membrane structure preceding cell lysis . J Immunol 2008 ; 181 : 822 - 832
Byrd JC, Kitada S, Flinn IW, Aron JL, Pearson M, Lucas D, Reed JC . The mechanism of tumor cell clearance by rituximab in vivo in patients with B-cell chronic lymphocytic leukemia: evidence of caspase activation and apoptosis induction . Blood 2002 ; 99 : 1038 - 1043
Carter PJ, Senter PD . Antibody-drug conjugates for cancer therapy . Cancer J 2008 ; 14 : 154 - 169
Cassinotti A, Ardizzone S, Porro GB . Certolizumab pegol: an evidence-based review of its place in the treatment of Crohn's disease . Core Evid 2008 ; 2 : 209 - 229
Centocor . Simponi full prescribing information . 2009, https://www.simponi.com/sites/default/files/pdf/prescribing-information.pdf
Chung KY, Shia J, Kemeny NE, Shah M, Schwartz GK, Tse A, Hamilton A, Pan D, Schrag D, Schwartz L, Klimstra DS, Fridman D, Kelsen DP, Saltz LB . Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry . J Clin Oncol 2005 ; 23 : 1803 - 1810
Clark M . Antibody humanization: a case of the 'Emperor's new clothes' ? Immunol Today 2000 ; 21 : 397 - 402
Clynes RA, Towers TL, Presta LG, Ravetch JV . Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets . Nat Med 2000 ; 6 : 443 - 446
Cohenuram M, Saif MW . Panitumumab the first fully human monoclonal antibody: from the bench to the clinic . Anticancer Drugs 2007 ; 18 : 7 - 15
Cooley S, Burns LJ, Repka T, Miller JS . Natural killer cell cytotoxicity of breast cancer targets is enhanced by two distinct mechanisms of antibody-dependent cellular cytotoxicity against LFA-3 and HER2/neu . Exp Hematol 1999 ; 27 : 1533 - 1541
Cragg MS, Morgan SM, Chan HTC, Morgan BP, Filatov AV, Johnson PW, French RR, Glennie MJ . Complement-mediated lysis by anti-CD20 mAb correlates with segregation into lipid rafts . Blood 2003 ; 101 : 1045 - 1052
Cragg MS, Glennie MJ . Antibody specificity controls in vivo effector mechanisms of anti-CD20 reagents . Blood 2004 ; 103 : 2738 - 2743
Craigen JL, Mackus WJM, Engleberts P, Miller SR, Speller S, Chamberlain LC, Davis BG, McHugh SM, Bullmore E, Cox CJ, Wetten S, Perdock G, Bakker JM, van de Winkel JGJ, Parren PWHI . Ofatumumab, a human mab targeting a membrane-proximal small-loop epitope on CD20, induces potent NK cell-mediated ADCC . Blood (ASH annual meeting abstracts) 2009 ; 114 : 1725
Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, Chau I, Van Cutsem E . Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer . N Engl J Med 2004 ; 351 : 337 - 345
Demidem A, Lam T, Alas S, Hariharan K, Hanna N, Bonavida B . Chimeric anti-CD20 (IDEC-C2B8) monoclonal antibody sensitizes a B cell lymphoma cell line to cell killing by cytotoxic drugs . Cancer Biother Radiopharm 1997 ; 12 : 177 - 186
Dunn EF, Iida M, Myers RA, Campbell DA, Hintz KA, Armstrong EA, Li C, Wheeler DL . Dasatinib sensitizes KRAS mutant colorectal tumors to cetuximab . Oncogene 2011 ; 30 : 561 - 574
Einfeld DA, Brown JP, Valentine MA, Clark EA, Ledbetter JA . Molecular cloning of the human B cell CD20 receptor predicts a hydrophobic protein with multiple transmembrane domains . Embo J 1988 ; 7 : 711 - 717
Emi Aikawa N, de Carvalho JF, Artur Almeida, Bonfa E . Immunogenicity of anti-TNF-alpha agents in autoimmune diseases . Clin Rev Allergy Immunol 2010 ; 38 : 82 - 89
Feldmann M, Brennan FM, Maini RN . Role of cytokines in rheumatoid arthritis . Annu Rev Immunol 1996 ; 14 : 397 - 440
Flieger D, Renoth S, Beier I, Sauerbruch T, Schmidt-Wolf I . Mechanism of cytotoxicity induced by chimeric mouse human monoclonal antibody IDEC-C2B8 in CD20-expressing lymphoma cell lines . Cell Immunol 2000 ; 204 : 55 - 63
Franklin MC, Carey KD, Vajdos FF, Leahy DJ, de Vos AM, Sliwkowski MX . Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex . Cancer Cell 2004 ; 5 : 317 - 328
Freeman D, Sun J, Bass R, Jung K, Ogbagabriel S, Elliott G, Radinsky R . Panitumumab and cetuximab epitope mapping and in vitro activity . J Clin Oncol 2008 ; 26 : (20 suppl) abstr 14536
Gebbia V, Giuliani F, Valori VM, Agueli R, Coiucci G, Maiello E . Cetuximab in squamous cell head and neck carcinomas . Ann Oncol 2007 ; 18 : 5 - 7
Genentech . Rituxan full prescribing information . 1997, http://www.gene.com/gene/products/information/pdf/rituxan-prescribing.pdf
Genentech . U.S. BLA Supplement: HERCEPTIN . 2002, http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/ucm092753.pdf
Gennari R, Menard S, Fagnoni F, Ponchio L, Scelsi M, Tagliabue E, Castiglioni F, Villani L, Magalotti C, Gibelli N, Oliviero B, Ballardini B, Da Prada G, Zambelli A, Costa A . Pilot study of the mechanism of action of preoperative trastuzumab in patients with primary operable breast tumors overexpressing HER2 . Clin Cancer Res 2004 ; 10 : 5650 - 5655
Ghosh R, Narasanna A, Wang SE, Liu S, Chakrabarty A, Balko JM, Gonzalez-Angulo AM, Mills GB, Penuel E, Winslow J, Sperinde J, Dua R, Pidaparthi S, Mukherjee A, Leitzel K, Kostler WJ, Lipton A, Bates M, Arteaga CL . Trastuzumab has preferential activity against breast cancers driven by HER2 homodimers . Cancer Res 2011 ; 71 : 1871 - 1882
Goldstein NI, Prewett M, Zuklys K, Rockwell P, Mendelsohn J . Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model . Clin Cancer Res 1995 ; 1 : 1311 - 1318
Heinemann V, Stintzing S, Kirchner T, Boeck S, Jung A . Clinical relevance of EGFR- and KRAS-status in colorectal cancer patients treated with monoclonal antibodies directed against the EGFR . Cancer Treat Rev 2009 ; 35 : 262 - 271
Huang SM, Li J, Harari PM . Molecular inhibition of angiogenesis and metastatic potential in human squamous cell carcinomas after epidermal growth factor receptor blockade . Mol Cancer Ther 2002 ; 1 : 507 - 514
Hudis CA . Trastuzumab - mechanism of action and use in clinical practice . N Engl J Med 2007 ; 357 : 39 - 51
Hynes NE, MacDonald G . ErbB receptors and signaling pathways in cancer . Curr Opin Cell Biol 2009 ; 21 : 177 - 184
Izumi Y, Xu L, di Tomaso E, Fukumura D, Jain RK . Tumour biology: Herceptin acts as an anti-angiogenic cocktail . Nature 2002 ; 416 : 279 - 280
Jakobovits A, Amado RG, Yang X, Roskos L, Schwab G . From XenoMouse technology to panitumumab, the first fully human antibody product from transgenic mice . Nat Biotechnol 2007 ; 25 : 1134 - 1143
Junttila TT, Akita RW, Parsons K, Fields C, Lewis Phillips GD, Friedman LS, Sampath D, Sliwkowski MX . Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941 . Cancer Cell 2009 ; 15 : 429 - 440
Kaneko E, Niwa R . Optimizing therapeutic antibody function: progress with Fc domain engineering . BioDrugs 2011 ; 25 : 1 - 11
Kawaguchi Y, Kono K, Mimura K, Sugai H, Akaike H, Fujii H . Cetuximab induce antibody-dependent cellular cytotoxicity against EGFR-expressing esophageal squamous cell carcinoma . Int J Cancer 2007 ; 120 : 781 - 787
Kim GP, Grothey A . Targeting colorectal cancer with human anti-EGFR monoclonocal antibodies: focus on panitumumab . Biologics 2008 ; 2 : 223 - 228
Kim MS, Lee SH, Song MY, Yoo TH, Lee BK, Kim YS . Comparative analyses of complex formation and binding sites between human tumor necrosis factor-alpha and its three antagonists elucidate their different neutralizing mechanisms . J Mol Biol 2007 ; 374 : 1374 - 1388
Kiyota A, Shintani S, Mihara M, Nakahara Y, Ueyama Y, Matsumura T, Tachikawa T, Wong DT . Anti-epidermal growth factor receptor monoclonal antibody 225 upregulates p27(KIP1) and p15(INK4B) and induces G1 arrest in oral squamous carcinoma cell lines . Oncology 2002 ; 63 : 92 - 98
Klos KS, Zhou X, Lee S, Zhang L, Yang W, Nagata Y, Yu D . Combined trastuzumab and paclitaxel treatment better inhibits ErbB-2-mediated angiogenesis in breast carcinoma through a more effective inhibition of Akt than either treatment alone . Cancer 2003 ; 98 : 1377 - 1385
Köhler G, Milstein C . Continuous cultures of fused cells secreting antibody of predefined specificity . Nature 1975 ; 256 : 495 - 497
Kohno T, Tam LT, Stevens SR, Louie JS . Binding characteristics of tumor necrosis factor receptor-Fc fusion proteins vs anti-tumor necrosis factor mAbs . J Investig Dermatol Symp Proc 2007 ; 12 : 5 - 8
Kristensen LE, Christensen R, Bliddal H, Geborek P, Danneskiold-Samsoe B, Saxne T . The number needed to treat for adalimumab, etanercept, and infliximab based on ACR50 response in three randomized controlled trials on established rheumatoid arthritis: a systematic literature review . Scand J Rheumatol 2007 ; 36 : 411 - 417
Kurai J, Chikumi H, Hashimoto K, Yamaguchi K, Yamasaki A, Sako T, Touge H, Makino H, Takata M, Miyata M, Nakamoto M, Burioka N, Shimizu E . Antibody-dependent cellular cytotoxicity mediated by cetuximab against lung cancer cell lines . Clin Cancer Res 2007 ; 13 : 1552 - 1561
Kute T, Lack CM, Willingham M, Bishwokama B, Williams H, Barrett K, Mitchell T, Vaughn JP . Development of herceptin resistance in breast cancer cells . Cytometry A 2004 ; 57 : 86 - 93
Launois R, Avouac B, Berenbaum F, Blin O, Bru I, Fautrel B, Joubert JM, Sibilia J, Combe B . Comparison of certolizumab pegol with other anticytokine agents for treatment of rheumatoid arthritis: A multiple-treatment Bayesian metaanalysis . J Rheumatol 2011 ; 38 : 835 - 845
Lee J, Lee I, Han B, Park JO, Jang J, Park C, Kang WK . Effect of simvastatin on cetuximab resistance in human colorectal cancer with KRAS mutations . J Natl Cancer Inst 2011 ; 103 : 674 - 688
Lewis GD, Figari I, Fendly B, Wong WL, Carter P, Gorman C, Shepard HM . Differential responses of human tumor cell lines to anti-p185HER2 monoclonal antibodies . Cancer Immunol Immunother 1993 ; 37 : 255 - 263
Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM . Structural basis for inhibition of the epidermal growth factor receptor by cetuximab . Cancer Cell 2005 ; 7 : 301 - 311
Lièvre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Cote JF, Tomasic G, Penna C, Ducreux M, Rougier P, Penault-Llorca F, Laurent-Puig P . KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer . Cancer Res 2006 ; 66 : 3992 - 3995
Mahtani RL, Macdonald JS . Synergy between cetuximab and chemotherapy in tumors of the gastrointestinal tract . Oncologist 2008 ; 13 : 39 - 50
Maini RN, Breedveld FC, Kalden JR, Smolen JS, Davis D, Macfarlane JD, Antoni C, Leeb B, Elliott MJ, Woody JN, Schaible TF, Feldmann M . Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor alpha monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis . Arthritis Rheum 1998 ; 41 : 1552 - 1563
Mandal M, Adam L, Mendelsohn J, Kumar R . Nuclear targeting of Bax during apoptosis in human colorectal cancer cells . Oncogene 1998 ; 17 : 999 - 1007
McLaughlin P, Grillo-Lopez AJ, Link BK, Levy R, Czuczman MS, Williams ME, Heyman MR, Bence-Bruckler I, White CA, Cabanillas F, Jain V, Ho AD, Lister J, Wey K, Shen D, Dallaire BK . Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: Half of patients respond to a four-dose treatment program . J Clin Oncol 1998 ; 16 : 2825 - 2833
Meira DD, Nobrega I, de Almeida VH, Mororo JS, Cardoso AM, Silva RL, Albano RM, Ferreira CG . Different antiproliferative effects of matuzumab and cetuximab in A431 cells are associated with persistent activity of the MAPK pathway . Eur J Cancer 2009 ; 45 : 1265 - 1273
Messersmith WA, Hidalgo M . Panitumumab, a monoclonal anti epidermal growth factor receptor antibody in colorectal cancer: another one or the one ? Clin Cancer Res 2007 ; 13 : 4664 - 4666
Molina MA, Codony-Servat J, Albanell J, Rojo F, Arribas J, Baselga J . Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells . Cancer Res 2001 ; 61 : 4744 - 4749
Müller D, Kontermann RE . Bispecific antibodies for cancer immunotherapy: Current perspectives . BioDrugs 2010 ; 24 : 89 - 98
Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, Laccabue D, Zerbini A, Camisa R, Bisagni G, Neri TM, Ardizzoni A . Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer . J Clin Oncol 2008 ; 26 : 1789 - 1796
Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, Hortobagyi GN, Hung MC, Yu D . PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients . Cancer Cell 2004 ; 6 : 117 - 127
Nahta R, Hung MC, Esteva FJ . The HER-2-targeting antibodies trastuzumab and pertuzumab synergistically inhibit the survival of breast cancer cells . Cancer Res 2004 ; 64 : 2343 - 2346
Nesbitt A, Fossati G, Bergin M, Stephens P, Stephens S, Foulkes R, Brown D, Robinson M, Bourne T . Mechanism of action of certolizumab pegol (CDP870): in vitro comparison with other anti-tumor necrosis factor alpha agents . Inflamm Bowel Dis 2007 ; 13 : 1323 - 1332
Nicholson RI, Gee JM, Harper ME . EGFR and cancer prognosis . Eur J Cancer 2001 ; 37 : S9 - S15
Palframan R, Airey M, Moore A, Vugler A, Nesbitt A . Use of biofluorescence imaging to compare the distribution of certolizumab pegol, adalimumab, and infliximab in the inflamed paws of mice with collagen-induced arthritis . J Immunol Methods 2009 ; 348 : 36 - 41
Pedersen IM, Buhl AM, Klausen P, Geisler CH, Jurlander J . The chimeric anti-CD20 antibody rituximab induces apoptosis in B-cell chronic lymphocytic leukemia cells through a p38 mitogen activated protein-kinase-dependent mechanism . Blood 2002 ; 99 : 1314 - 1319
Perrotte P, Matsumoto T, Inoue K, Kuniyasu H, Eve BY, Hicklin DJ, Radinsky R, Dinney CP . Anti-epidermal growth factor receptor antibody C225 inhibits angiogenesis in human transitional cell carcinoma growing orthotopically in nude mice . Clin Cancer Res 1999 ; 5 : 257 - 265
Persson M, Tolmachev V, Andersson K, Gedda L, Sandstrom M, Carlsson J . [(177)Lu]pertuzumab: experimental studies on targeting of HER-2 positive tumour cells . Eur J Nucl Med Mol Imaging 2005 ; 32 : 1457 - 1462
Petit AM, Rak J, Hung MC, Rockwell P, Goldstein N, Fendly B, Kerbel RS . Neutralizing antibodies against epidermal growth factor and ErbB-2/neu receptor tyrosine kinases down-regulate vascular endothelial growth factor production by tumor cells in vitro and in vivo: angiogenic implications for signal transduction therapy of solid tumors . Am J Pathol 1997 ; 151 : 1523 - 1530
Press OW, Appelbaum F, Ledbetter JA, Martin PJ, Zarling J, Kidd P, Thomas ED . Monoclonal antibody 1F5 (anti-CD20) serotherapy of human B cell lymphomas . Blood 1987 ; 69 : 584 - 591
Ramakrishnan MS, Eswaraiah A, Crombet T, Piedra P, Saurez G, Iyer H, Arvind AS . Nimotuzumab, a promising therapeutic monoclonal for treatment of tumors of epithelial origin . MAbs 2009 ; 1 : 41 - 48
Ritchlin C, Haas-Smith SA, Hicks D, Cappuccio J, Osterland CK, Looney RJ . Patterns of cytokine production in psoriatic synovium . J Rheumatol 1998 ; 25 : 1544 - 1552
Rivkin A . Certolizumab pegol for the management of Crohn's disease in adults . Clin Ther 2009 ; 31 : 1158 - 1176
Rockberg J, Lofblom J, Hjelm B, Uhlen M, Stahl S . Epitope mapping of antibodies using bacterial surface display . Nature Methods 2008 ; 5 : 1039 - 1045
Rockberg J, Schwenk JM, Uhlen M . Discovery of epitopes for targeting the human epidermal growth factor receptor 2 (HER2) with antibodies . Mol Oncol 2009 ; 3 : 238 - 247
Rudnick SI, Lou J, Shaller CC, Tang Y, Klein-Szanto AJ, Weiner LM, Marks JD, Adams GP . Influence of affinity and antigen internalization on the uptake and penetration of Anti-HER2 antibodies in solid tumors . Cancer Res 2011 ; 71 : 2250 - 2259
Saltz LB, Meropol NJ, Loehrer PJ, Needle MN, Kopit J, Mayer RJ . Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor . J Clin Oncol 2004 ; 22 : 1201 - 1208
Schmitz KR, Ferguson KM . Interaction of antibodies with ErbB receptor extracellular regions . Exp Cell Res 2009 ; 315 : 659 - 670
Schneider-Merck T, Lammerts van, Berger S, Rossen K, van Berkel PH, Derer S, Beyer T, Lohse S, Bleeker WK, Peipp M, Parren PW, van de Winkel JG, Valerius T, Dechant M . Human IgG2 antibodies against epidermal growth factor receptor effectively trigger antibody-dependent cellular cytotoxicity but, in contrast to IgG1, only by cells of myeloid lineage . J Immunol 2010 ; 184 : 512 - 520
Shealy D, Cai A, Staquet K, Baker A, Lacy ER, Johns L, Vafa O, Gunn G, Tam S, Sague S, Wang D, Brigham-Burke M, Dalmonte P, Emmell E, Pikounis B, Bugelski PJ, Zhou H, Scallon B, Giles-Komar J . Characterization of golimumab, a human monoclonal antibody specific for human tumor necrosis factor alpha . MAbs 2010 ; 2
Skvortsova I, Skvortsov S, Popper BA, Haidenberger A, Saurer M, Gunkel AR, Zwierzina H, Lukas P . Rituximab enhances radiation-triggered apoptosis in non-Hodgkin's lymphoma cells via caspase-dependent and - independent mechanisms . J Radiat Res (Tokyo) 2006 ; 47 : 183 - 196
Spiridon CI, Guinn S, Vitetta ES . A comparison of the in vitro and in vivo activities of IgG and F(ab')2 fragments of a mixture of three monoclonal anti-Her-2 antibodies . Clin Cancer Res 2004 ; 10 : 3542 - 3551
Teeling JL, French RR, Cragg MS, van den Brakel J, Pluyter M, Huang H, Chan C, Parren PW, Hack CE, Dechant M, Valerius T, van de Winkel JG, Glennie MJ . Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas . Blood 2004 ; 104 : 1793 - 1800
Teeling JL, Mackus WJ, Wiegman LJ, Van den Brakel JH, Beers SA, French RR, van Meerten T, Ebeling S, Vink T, Slootstra JW, Parren PW, Glennie MJ, Van de Winkel JG . The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20 . J Immunol 2006 ; 177 : 362 - 371
UCB . Cimzia full prescribing information . 2008, http://www.ucb.com/_up/ucb_com_products/documents/Cimzia_12_21_10.pdf
Uchiyama S, Suzuki Y, Otake K, Yokoyama M, Ohta M, Aikawa S, Komatsu M, Sawada T, Kagami Y, Morishima Y, Fukui K . Development of novel humanized anti-CD20 antibodies based on affinity constant and epitope . Cancer Sci 2010 ; 101 : 201 - 209
Valabrega G, Montemurro F, Aglietta M . Trastuzumab: mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer . Ann Oncol 2007 ; 18 : 977 - 984
Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF, Burchmore M, Shak S, Stewart SJ, Press M . Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer . J Clin Oncol 2002 ; 20 : 719 - 726
Williams RO, Paleolog E, Feldmann M . Cytokine inhibitors in rheumatoid arthritis and other autoimmune diseases . Curr Opin Pharmacol 2007 ; 7 : 412 - 417
Wu X, Rubin M, Fan Z, DeBlasio T, Soos T, Koff A, Mendelsohn J . Involvement of p27KIP1 in G1 arrest mediated by an anti-epidermal growth factor receptor monoclonal antibody . Oncogene 1996 ; 12 : 1397 - 1403
Yang XD, Jia XC, Corvalan JR, Wang P, Davis CG, Jakobovits A . Eradication of established tumors by a fully human monoclonal antibody to the epidermal growth factor receptor without concomitant chemotherapy . Cancer Res 1999 ; 59 : 1236 - 1243
Yoon S, Kim YS, Shim H, Chung J . Current perspectives on therapeutic antibodies . Biotechnol Bioprocess Eng 2010 ; 15 : 709 - 715
zum Buschenfelde CM, Hermann C, Schmidt B, Peschel C, Bernhard H . Antihuman epidermal growth factor receptor 2 (HER2) monoclonal antibody trastuzumab enhances cytolytic activity of class I-restricted HER2-specific T lymphocytes against HER2-overexpressing tumor cells . Cancer Res 2002 ; 62 : 2244 - 2247
Acknowledgements
This work was supported by the Global Frontier Project grant (NRF-M1AXA-002-2010-0029762) of National Research Foundation funded by the Ministry of Education, Science and Technology of Korea.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Shim, H. One target, different effects: a comparison of distinct therapeutic antibodies against the same targets. Exp Mol Med 43, 539–549 (2011). https://doi.org/10.3858/emm.2011.43.10.063
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2011.43.10.063
Keywords
This article is cited by
-
A review of ocular adverse events of biological anti-TNF drugs
Journal of Ophthalmic Inflammation and Infection (2020)
-
Affinity-matured variants derived from nimotuzumab keep the original fine specificity and exhibit superior biological activity
Scientific Reports (2020)
-
Soluble ligands as drug targets
Nature Reviews Drug Discovery (2020)
-
Anti-PD-1 monoclonal antibody MEDI0680 in a phase I study of patients with advanced solid malignancies
Journal for ImmunoTherapy of Cancer (2019)
-
Anti-Toxoplasma antibodies in Egyptian rheumatoid arthritis patients
Rheumatology International (2017)
