
Abstract
Angiotensin II is a major effector molecule in the development of cardiovascular disease. In vascular smooth muscle cells (VSMCs), angiotensin II promotes cellular proliferation and extracellular matrix accumulation through the upregulation of plasminogen activator inhibitor-1 (PAI-1) expression. Previously, we demonstrated that small heterodimer partner (SHP) represses PAI-1 expression in the liver through the inhibition of TGF-β signaling pathways. Here, we investigated whether SHP inhibited angiotensin II-stimulated PAI-1 expression in VSMCs. Adenovirus-mediated overexpression of SHP (Ad-SHP) in VSMCs inhibited angiotensin II- and TGF-β-stimulated PAI-1 expression. Ad-SHP also inhibited angiotensin II-, TGF-β- and Smad3-stimulated PAI-1 promoter activity, and angiotensin II-stimulated AP-1 activity. The level of PAI-1 expression was significantly higher in VSMCs of SHP-/- mice than wild type mice. Moreover, loss of SHP increased PAI-1 mRNA expression after angiotensin II treatment. These results suggest that SHP inhibits PAI-1 expression in VSMCs through the suppression of TGF-β/Smad3 and AP-1 activity. Thus, agents that target the induction of SHP expression in VSMCs might help prevent the development and progression of atherosclerosis.
Similar content being viewed by others

Introduction
Plasminogen activator inhibitor-1 (PAI-1) was identified as a significant biomarker and predictor of cardiovascular disease-related death in the Framingham Heart Study (Wang et al., 2006), and several studies have reported an important role for PAI-1 in the development of arteriosclerosis and perivascular fibrosis (Sobel et al., 2003; Weisberg et al., 2005; Vaughan et al., 2006). Moreover, in addition to circulating PAI-1, arterial PAI-1 expression is increased by elevated angiotensin II in patients with hypertension (Vaughan et al., 1995; Kaikita et al., 2001).
Angiotensin II is a major effector molecule in the renin-angiotensin system. It regulates systemic blood pressure through a direct vasopressive effect, and through the induction of vasoactive compounds (Ito et al., 1995; Ferrario et al.,1998; Weir and Dzau, 1999). Previous findings suggest that angiotensin II promotes cellular proliferation and extracellular matrix accumulation in vascular smooth muscle cells (VSMCs) through the upregulation of PAI-1 expression (Kato et al., 1991; van Leeuwen et al., 1994; Papakonstantinou et al., 2001), and several transcription factors, including Smad3 and AP-1, have been implicated as important mediators of angiotensin II-stimulated PAI-1 expression in VSMCs (Ahn et al., 2001; Binder et al., 2002; Wang et al., 2006). Thus, clarifying the signaling networks that underlie angiotensin II-induced PAI-1 expression might reveal novel, therapeutically useful targets for managing angiotensin II/PAI-1-dependent cardiovascular disorders.
The orphan nuclear receptor SHP is an atypical member of the orphan nuclear receptor superfamily because it lacks a conventional DNA binding domain (Seol et al., 1996). SHP is a transcriptional repressor that exerts its regulatory effects through protein-protein interactions with other nuclear hormone receptors, and possibly other transcription factors, inhibiting or reversing transactivation (Seol et al., 1998; Gobinet et al., 2001; Brendel et al., 2002; Lee and Moore, 2002; Sanyal et al., 2002; Ourlin et al., 2003; Yamagata et al., 2004). Previously, our group demonstrated that SHP represses hepatic PAI-1 expression through the inhibition of TGF-β signaling pathways and the repression of Smad3 transactivation (Suh et al., 2006). These results suggested that direct targeting of SHP might be a promising approach to the prevention of atherosclerosis, particularly in patients with hypertension. However, little is known about the role of SHP in vascular cell function, particularly in angiotensin II-stimulated PAI-1 gene expression. Here, we examined the effect of SHP on angiotensin II-stimulated PAI-1 expression in primary cultured rat VSMCs, and elucidated a putative molecular mechanism of action of SHP in these cells.
Results
SHP inhibits angiotensin II-stimulated PAI-1 expression in primary cultured rat VSMCs
To investigate whether SHP inhibited PAI-1 expression in primary cultured rat VSMCs, we measured the effect of adenovirus-mediated overexpression of SHP in VSMCs on angiotensin II-stimulated PAI-1 mRNA expression by Northern blot analysis. As shown in Figure 1A, Ad-SHP decreased angiotensin II-stimulated PAI-1 expression in a dose-dependent manner. Transient transfection experiments using a luciferase reporter gene under the control of the PAI-1 promoter revealed that SHP expression decreased PAI-1 promoter activity in a dose-dependent manner (Figure 1B). Additionally, transient SHP expression decreased angiotensin II-stimulated PAI-1 promoter activity in a dose-dependent manner (Figure 1C). These data suggested that the inhibition of angiotensin II-stimulated PAI-1 gene expression by SHP is mediated at the transcriptional level.

Effect of SHP on angiotensin II-stimulated PAI-1 mRNA expression and promoter activity. (A) Northern blot analysis of the effect of overexpression of SHP on PAI-1 mRNA expression. VSMCs were infected at the indicated multiplicity-of-infection (moi) with adenovirus expressing SHP (Ad-SHP), or green fluorescent protein (GFP) as a control, for 4 h. VSMCs were incubated in medium containing 0.5% FBS for 24 h, and then treated with angiotensin II (Ang II, 10 nmol/l) for 3 h before harvest. Data represent the means ± SEM. *P < 0.05, **P < 0.01, #P < 0.001 as compared to Ang II alone. (B and C) Luciferase gene reporter assay of the effect of SHP on PAI-1 promoter activity. (B) VSMCs were transfected with a reporter gene under the control of the PAI-1 promoter (-840 PAI-1 promoter Luc, 300 ng) together with the indicated amounts of an SHP expression vector (pcDNA3) for 5 h, and were then incubated in medium containing 10% FBS for 24h. Data represent the means ± SEM. *P < 0.01, **P < 0.001 were as compared with the control. (C) VSMCs were transfected with a reporter gene under the control of the PAI-1 promoter (-840 PAI-1 promoter Luc, 300 ng) together with the indicated amounts of an SHP expression vector (pcDNA3) for 5 h. Cells were incubated in medium containing 0.5% FBS for 24 h, and then treated with angiotensin II (Ang II, 10 nM) for 1 h before harvest. Data represent the means ± SEM. *P < 0.001 as compared to control. **P < 0.01, #P < 0.001 as compared to Ang II alone.
SHP inhibits PAI-1 expression through the down-regulation of the TGF-β/Smad3 pathway
PAI-1 activity is tightly regulated at the transcriptional level (Binder et al., 2002). Given that TGF-β is an important regulator of angiotensin II-induced PAI-1 transcription (Rodriguez-Vita et al., 2005; Ruiz-Ortega et al., 2007), we examined whether Ad-SHP inhibited TGF-β-stimulated PAI-1 transcription. Infection of cells with Ad-SHP significantly inhibited TGF-β-stimulated PAI-1 mRNA expression in a dose-dependent manner (Figure 2A). Similarly, transient expression of SHP in VSMCs significantly inhibited TGF-β-stimulated PAI-1 promoter activity in a dose-dependent manner (Figure 2B upper). Transient expression of SHP also significantly inhibited Smad3-stimulated PAI-1 promoter activity in a dose-dependent manner (Figure 2B lower). Taken together, these data suggested that SHP inhibits PAI-1 expression through the inhibition of the TGF-β/Smad3 signaling pathway.

Effect of SHP on TGF-β-stimulated PAI-1 mRNA expression and Smad3 activity. (A) Northern blot analysis of the effect of overexpression of SHP on PAI-1 m RNA expression. VSMCs were infected at the indicated moi with Ad-SHP or GFP for 4 h. Cells were incubated in medium containing 0.5% FBS 24 h, and then treated with TGF-β (1 ng/ml) for 3 h before harvest. Data represents the means ± SEM. *P < 0.05, **P < 0.01 as compared to TGF-β alone. (B) Luciferase gene reporter assay of the effect of SHP on PAI-1 promoter activity. (Upper) VSMCs were transfected with -840 PAI-1 promoter Luc (300 ng) together with the indicated amounts of an SHP expression vector (pcDNA3) for 5 h. Cells were incubated in medium containing 0.5% FBS for 24 h, and then treated with TGF-β (1 ng/ml) for 1 h before harvest. Data represents the means ± SEM. *P < 0.01 as compared to the control. **P < 0.05, #P < 0.01 as compared to TGF-β alone. (Lower) VSMCs were co-transfected with -840 PAI-1 promoter Luc (300 ng) and expression vectors for Smad3 (100 ng), Smad4 (100 ng) and ALK5 (100 ng), together with the indicated amounts of SHP expression vector for 5 h. Cells were then incubated in medium containing 0.5% FBS for 24 h. Data represents the means ± SEM. *P < 0.001 as compared to control. **P < 0.01, #P < 0.001 as compared to Smad3/4/ALK5 alone.
SHP inhibits angiotensin-II-stimulated AP-1 activity
To determine whether SHP inhibited angiotensin II-stimulated PAI-1 transcription through a TGF-β/Smad3 independent pathway, VSMCs were co-transfected with a luciferase reporter gene under the control of a truncated form of the PAI-1 promoter, -240 PAI-1, in which two Smad binding sites were deleted (Figure 3A). Angiotensin II treatment stimulated the activity of the truncated promoter construct, although the level of activity was lower than that of the WT promoter construct (Figure 3A). These results suggested that in addition to Smad, other factors contribute to angiotensin II-stimulated PAI-1 expression. SHP inhibited angiotensin II-mediated -240 PAI-1 promoter activity, reinforcing the idea that SHP inhibits transcription factors other than Smad (Figure 3A). We also examined the effect of SHP on the activity of AP-1, which is the primary transcription factor involved in stimulating PAI-1 promoter activity in response to angiotensin II. AP-1 DNA binding was stimulated by angiotensin II (10 nmol/l), and adenovirus-mediated overexpression of SHP inhibited angiotensin II-stimulated AP-1 binding activity in a dose-dependent manner (Figure 3B).

Effect of SHP on angiotensin II-stimulated AP-1 activity. (A) Luciferase gene reporter assay of the effect of SHP on Smad3-independent PAI-1 promoter activity. VSMCs were transfected with a luciferase reporter gene under the control of an SBE deleted PAI-1 promoter (-240 PAI-1 promoter Luc, 300 ng), together with the indicated amounts of SHP expression vector for 5 h. Cells were incubated in medium containing 0.5% FBS for 24 h, and then treated with angiotensin II (Ang II, 10 nM) for 1 h before harvest. Data represents the means ± SEM. *P < 0.001 as compared to control. **P < 0.01 as compared to Ang II alone (WT PAI-1 promoter), #P < 0.05 as compared to Ang II alone (SBE-deleted PAI-1 promoter). (B) Electrophoretic mobility shift assay of the effect of SHP on AP-1 DNA binding activity. VSMCs were infected at the indicated moi with Ad-SHP or GFP for 4 h. Cells were incubated in medium containing 0.5% FBS for 24 h, and then treated with angiotensin II (Ang II, 10 nmol/l) for 1 h before harvest. (C) VSMCs were cotransfected with the -840 PAI-1 promoter Luc (300 ng) and the AP-1 decoy ODN (10 nM) or mismatched AP-1 decoy ODN (MD, 10 nM) together with the indicated amounts of an SHP expression vector (pcDNA3) or c-jun/c-fos expression vector for 5 h. Cells were incubated in medium containing 0.5% FBS for 24 h, and were then treated with angiotensin II (Ang II, 10 nmol/l) for 1 h before harvesting. Data represents the means ± SEM. *P < 0.05, ##P < 0.001 as compared to the control. **P < 0.05, #P < 0.01 were as compared to Ang II.
To see whether SHP inhibits angiotensin II-stimulated PAI-1 promoter activity independently from AP-1 inhibition, AP-1 decoy ODN, which inhibits the AP-1 binding to the PAI-1 promoter, was used. Angiotensin II stimulated the activity of the -840 PAI-1 promoter treated with AP-1 decoy OND, although the level of activity was lower than that of the -840 PAI-1 promoter treated with mismatched AP-1 ODN. SHP inhibited angiotensin II-stimulated PAI-1 promoter activity in a dose dependent manner. AP-1 decoy efficiency was confirmed by transient transfection with AP-1 proteins. c-jun and c-fos did not stimulate the activity of the -840 PAI-1 promoter treated with AP-1 decoy ODN (Figure 3C). Collectively, these data suggested that SHP inhibits PAI-1 expression in VSMCs through the downregulation of TGF-β/Smad3 and AP-1 activity.
PAI-1 mRNA expression is upregulated in primary cultured VSMCs of SHP-/- mice
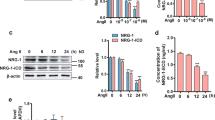
We next examined whether the loss of SHP influenced the expression of PAI-1 in VSMCs in SHP-/- mice. In primary cultured VSMCs from SHP-/- mice, PAI-1 mRNA levels were increased as compared to VSMCs of WT mice. Moreover, loss of SHP increased PAI-1 mRNA expression after angiotensin II treatment (Figure 4). These data suggested that endogenous SHP acts as negative regulator of PAI-1 expression in VSMCs in vivo.

PAI-1 expression in primary cultured VSMCs of SHP-/- mice. RT-PCR analysis of PAI-1 and SHP mRNA expression in primary cultured VSMCs from WT (SHP+/+) and SHP-knockout (SHP-/-) mice. Cells were incubated in medium containing 0.5% FBS for 24 h, and were then treated with angiotensin II (Ang II, 10 nM) for 3 h before harvesting. Data represent the means ± SEM. *P < 0.05 was as compared to WT VSMC (SHP+/+) without Ang II, **P < 0.05 was as compared to KO VSMC (SHP-/-) without Ang II, and #P < 0.05 was as compared to WT VSMC (SHP+/+) with Ang II.
Discussion
In the present study, we demonstrated that adenovirus-mediated overexpression of SHP decreases angiotensin II- and TGF-β-stimulated PAI-1 mRNA expression in primary cultured rat VSMCs. We also elucidated a potential mechanism of downregulation of PAI-1 expression by SHP involving the inhibition of both TGF-β/Smad3 and AP-1 activity. PAI-1 plays a critical role in cardiovascular disease and is a key mediator in the process of vascular fibrosis (Lyon and Hsueh, 2003). It has long been thought that angiotensin II induces vascular fibrosis by stimulating TGF-β (Ruiz-Ortega et al., 2003; Wang et al., 2006). TGF-β mediates its fibrotic effects through the activation of receptor-associated Smads, including Smad2 and Smad3 (Dennler et al., 1998; Javelaud et al., 2004; Ruiz-Ortega et al., 2007). Recent studies have shown that angiotensin II activates an early Smad signaling pathway in VSMCs directly through ERK1/2 MAPK, as well as the classic late TGF-β signaling pathway (Ruiz-Ortega et al., 2003, 2007; Rodirguez-Vita et al., 2005; Wang et al., 2006). In the current study, adenovirus-mediated overexpression of SHP in VSMCs inhibited angiotensin II-stimulated PAI-1 expression. SHP also inhibited TGF-β-stimulated PAI-1 expression and transactivation of the PAI-1 promoter by Smad3.
There is increasing evidence that, in addition to the TGF-β/Smad pathway, other transcription factors, such as AP-1, are also important in angiotensin II-induced PAI-1 expression (Ahn et al., 2001). In the current study, angiotensin II stimulated the activity of a luciferase reporter gene under the control of an SBE-deleted PAI-1 promoter, in which two Smad binding sites were deleted. These results suggest the involvement of transcription factors other than Smad3 in the action of angiotensin II. There is ample evidence that risk factors for atherosclerosis stimulate the transactivity of AP-1, a redox sensitive transcription factor, at the PAI-1 promoter. Previously, we and others have shown that angiotensin II increases PAI-1 expression through the upregulation of AP-1 activity in VSMCs (Ahn et al. 2001; Chen and Feener, 2004).
Previous reports have demonstrated the mechanisms by which SHP regulates target gene transcription without directly binding to target promoter sequences. SHP represses transcription factor-mediated transactivation by inhibiting DNA binding (Seol et al., 1996; Ourlin et al., 2003; Kim et al., 2004), by recruiting unknown corepressors (Johansson et al., 2000; Lee et al., 2000, 2002) and by interfering with the interaction with coactivator (Suh et al., 2006). Fiorucci et al. (2004) showed that the inhibitory effect on AP-1 activity is mediated by a physical interaction between AP-1 and SHP that decreases AP-1 DNA binding. Our results, using an electrophoretic mobility shift assay, suggest that the inhibition of angiotensin II-stimulated PAI-1 expression by SHP involves in part the inhibition of AP-1-DNA binding at the PAI-1 promoter. Thus, the effect of SHP on vascular PAI-1 expression is likely to be multifactorial, involving the inhibition of Smad3 and AP-1 activity.
In conclusion, we have demonstrated that SHP inhibits PAI-1 expression by suppressing angiotensin II-stimulated transactivation of Smad3 and AP-1. Agents that increase SHP expression in vascular cells might help prevent the development and progression of atherosclerosis in patients with hypertension.
Methods
Materials
Recombinant human TGF-β1 was purchased from R&D Systems (Minneapolis, MN). Recombinant human angiotensin II was purchased from Calbiochem (San Diego, CA). Radiochemicals ([α-32P] dCTP, [γ-32P] ATP) were purchased from Perkin Elmer (Boston, MA).
Cell culture
VSMCs were isolated from the thoracic aorta of 4-week-old Sprague-Dawley (Samtako, Korea) rats weighing 100 g, and from SHP-/- mice, using the explant method. SHP-/- mice were a kind gift from Dr. David D. Moore (Baylor College of Medicine, Houston, TX). VSMCs culture was performed as described previously (Kim et al., 2007). Briefly, VSMCs were cultured in DMEM (Gibco BRL, Grand Island, NY) containing 20% FBS (Hyclone, Logan, UT). VSMC purity was assessed by positive staining with smooth muscle specific α-actin monoclonal antibodies (Santa Cruz, Santa Cruz, CA). Cells from the third and fifth passages were used for all experiments.
Northern blot analysis
Total RNA was isolated from VSMCs using Trizol Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Twenty-microgram aliquots of total RNA from each sample were used. The probes for SHP and PAI-1 were labeled with [α-32P] dCTP using a random-primer DNA-labeling system (Amersham Biosciences, Little Chalfont, UK). Northern analysis was performed as described previously (Ahn et al., 2001).
In vitro transient transfection and reporter assays
VSMCs were plated at a density 7 × 104 per well in a 12-well plate and cultured for 24 h. Cells were transiently transfected with a luciferase reporter gene under the control of the wild-type (WT) PAI-1 (-840 PAI-1 promoter) promoter, or a Smad binding element (SBE)-deleted PAI-1 (-240 PAI-1 promoter) promoter (300 ng/well), along with the indicated expression vectors, using Lipofectamine Plus reagent (Invitrogen). Cells were co-transfected with an expression plasmid for β-galactosidase as an internal control. Cells were transfected for 5 h, then washed to remove unincorporated plasmids and cultured in conditioned culture medium. Cells were harvested approximately 24 h after transfection and assayed for luciferase and β-galactosidase activity. Cell lysate (20 µl) was analyzed using the luciferase assay system, according to the manufacturer's instructions (Promega, Madison, WI), and luciferase activity was detected using a SIRUS Luminometer (Berthold, Pforzheim, Germany). Luciferase activity was normalized to β-galactosidase activity.
Construction of the AP-1 decoy
The construction of decoy oligodeoxynucleotides (ODN) was performed as described previously (Ahn et al., 2002). The sequences of the dumbbell-shaped decoy ODN directed against the AP-1 binding site and its mismatched AP-1 decoy ODN were constructed as described (Ahn et al., 2002) and were as follows (with consensus sequences underlined): AP-1 decoy ODN, 5'-GGATCCATGACTCAGAAGACGACACACGTCTTCTGAGTCAT-3; Mismatched AP-1 decoy ODN, 5'-GGATCCAAATCTCAGAAGACGACACACGTCTTCTGAGATTT-3'. The stability and cellular uptake of the decoy ODN were similar to those previously described (Ahn et al., 2002).
Electrophoretic mobility shift assay
Nuclear extracts were isolated from cells using Nuc-Buster™ Protein Extraction kit (Calbiochem), according to the manufacturer's instructions. After centrifugation, supernatants (nuclear extracts) were collected and protein concentration was measured using a protein assay kit (Bio-Rad, Richmond, CA). Nuclear extracts (6 µg) were incubated with approximately 60,000 cpm of a 32P-labeled AP-1 binding-site oligomer, 5'-CGCTTGATGACTCAGCCGGAA-3', for 20 min at 20℃.
RT-PCR
Total RNA was obtained from VSMCs using Trizol Reagent (Invitrogen). cDNA was synthesized using a first-strand cDNA synthesis kit (Fermentas, Hanover, MD) and 2 µg of total RNA, according to the manufacturer's instructions. PCR was carried out using Taq polymerase (Takara, Japan) and the following thermal cycling parameters: 94℃ for 5 min; 35 cycles of 94℃ for 1 min; 70℃ for 50 s; and 72℃ for 1 min. The following primers and thermal cycling parameters were used for mSHP: 5'-CCGCACCGCACCTGCAGGGAGGCCTT-3' (forward) and 5'-ACTCCAGGCAGCGCTGCAGCCACTGAA-3' (reverse); 94℃ for 5 min; and then 30 cycles of 94℃ for 50 s, 67℃ for 50 s and 72℃ for 2 min. The following primers were used for standard PCR of mPAI-1 and actin: for mPAI-1, 5'-CCTCATCCTGGGCCTGGTTCTGGTCT-3' (forward) and 5'-GGTTTTCCCCGCTGTGGTCATCTGC-3' (reverse); for actin, 5'-GGCATCGTCACCAACTGGGAC-3' (forward) and 5'-CGATTTCCCGCTCCGTGG-3' (reverse).
Statistical analysis
Results are expressed as the means ± standard error of the mean (SEM). Variance analysis with a subsequent Duncan's test was used to determine significant differences in multiple comparisons. A P value of < 0.05 was considered statistically significant. All experiments were performed at least 3 times.
Abbreviations
- PAI-1:
-
plasminogen activator inhibitor-1
- SHP:
-
small heterodimer partner
- VSMCs:
-
vascular smooth muscle cells
References
Ahn JD, Morishita R, Kaneda Y, Lee KU, Park JY, Jeon YJ, Song HS, Lee IK . Transcription factor decoy for activator protein-1 (AP-1) inhibits high glucose- and angiotensin II-induced type 1 plasminogen activator inhibitor (PAI-1) gene expression in cultured human vascular smooth muscle cells . Diabetologia 2001 ; 44 : 713 - 720
Ahn JD, Morishita R, Kaneda Y, Lee SJ, Kwon KY, Choi SY, Lee KU, Park JY, Moon IJ, Park JG, Yoshizumi M, Ouchi Y, Lee IK . Inhibitory effects of novel AP-1 decoy oligodeoxynucleotides on vascular smooth muscle cell proliferation in vitro and neointimal formation in vivo . Circ Res 2002 ; 90 : 1234 - 1236
Binder BR, Christ G, Gruber F, Grubic N, Hufnagl P, Krebs M, Mihaly J, Prager GW . Plasminogen Activator Inhibitor 1: Physiological and Pathophysiological Roles . News Physiol Sci 2002 ; 17 : 56 - 61
Brendel C, Schoonjans K, Botrugno OA, Treuter E, Auwerx J . The small heterodimer partner interacts with the liver X receptor α and represses its transcriptional activity . Mol Endocrinol 2002 ; 16 : 2065 - 2076
Chen HC, Feener EP . MEK1,2 response element mediates angiotensin II-stimulated plasminogen activator inhibitor-1 promoter activation . Blood 2004 ; 103 : 2636 - 2644
Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM . Direct binding of Smad3 and Smad4 to critical TGF-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene . EMBO J 1998 ; 17 : 3091 - 3100
Ferrario CM, Chappell MC, Dean RH, Iyer SN . Novel angiotensin peptides regulate blood pressure, endothelial function, and natriuresis . J Am Soc Nephrol 1998 ; 9 : 1716 - 1722
Fiorucci S, Antonelli E, Rizzo G, Renga B, Mencarelli A, Riccardi L, Orlandi S, Pellicciari R, Morelli A . The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis . Gastroenterology 2004 ; 127 : 1497 - 1512
Gobinet J, Auzou G, Nicolas JC, Sultan C, Jalaguier S . Characterization of the interaction between androgen receptor and a new transcriptional inhibitor, SHP . Biochemistry 2001 ; 40 : 15369 - 15377
Ito M, Oliverio MI, Mannon PJ, Best CF, Maeda N, Smithies O, Coffman TM . Regulation of blood pressure by the type 1A angiotensin II receptor gene . Proc Natl Acad Sci USA 1995 ; 92 : 3521 - 3525
Javelaud D, Mauviel A . Mammalian transforming growth factor-betas: Smad signaling and physio-pathological roles . Int J Biochem Cell Biol 2004 ; 36 : 1161 - 1165
Johansson L, Båvner A, Thomsen JS, Färnegårdh M, Gustafsson JA, Treuter E . The orphan nuclear receptor SHP utilizes conserved LXXLL-related motifs for interactions with ligand-activated estrogen receptors . Mol Cell Biol 2000 ; 20 : 1124 - 1133
Kaikita K, Fogo AB, Ma LJ, Schoenhard JA, Brown NJ, Vaughan DE . Plasminogen activator inhibitor-1 deficiency prevents hypertension and vascular fibrosis in response to long-term nitric oxide synthase inhibition . Circulation 2001 ; 104 : 839 - 844
Kato H, Suzuki H, Tajima S, Ogata Y, Tominaga T, Sato A, Saruta T . Angiotensin II stimulates collagen synthesis in cultured vascular smooth muscle cells . J Hypertens 1991 ; 9 : 17 - 22
Kim HS, Kim HJ, Park KG, Kim YN, Kwon TK, Park JY, Lee KU, Kim JG, Lee IK . Alpha-lipoic acid inhibits matrix metalloproteinase-9 expression by inhibiting NF-kappaB transcriptional activity . Exp Mol Med 2007 ; 39 : 106 - 113
Kim JY, Kim HJ, Kim KT, Park YY, Seong HA, Park KC, Lee IK, Ha H, Shong M, Park SC, Choi HS . Orphan nuclear receptor small heterodimer partner represses hepatocyte nuclear factor 3/Foxa transactivation via inhibition of its DNA binding . Mol Endocrinol 2004 ; 18 : 2880 - 2894
Lee YK, Dell H, Dowhan DH, Hadzopoulou-Cladaras M, Moore DD . The orphan nuclear receptor SHP inhibits hepatocyte nuclear factor 4 and retinoid X receptor transactivation: two mechanisms for repression . Mol Cell Biol 2000 ; 20 : 187 - 195
Lee YK, Moore DD . Dual mechanisms for repression of the monomeric orphan receptor liver receptor homologous protein-1 by the orphan small heterodimer partner . J Biol Chem 2002 ; 277 : 2463 - 2467
Lyon CJ, Hsueh WA . Effect of plasminogen activator inhibitor-1 in diabetes mellitus and cardiovascular disease . Am J Med 2003 ; 115 : 62S - 68S
Ourlin JC, Lasserre F, Pineau T, Fabre JM, Sa-Cunha A, Maurel P, Vilarem MJ, Pascussi JM . The small heterodimer partner interacts with the pregnane X receptor and represses its transcriptional activity . Mol Endocrinol 2003 ; 17 : 1693 - 1703
Papakonstantinou E, Roth M, Kokkas B, Papadopoulos C, Karakiulakis G . Losartan inhibits the angiotensin II-induced modifications on fibrinolysis and matrix deposition by primary human vascular smooth muscle cells . J Cardiovasc Pharmacol 2001 ; 38 : 715 - 728
Rodriguez-Vita J, Sanchez-Lopez E, Esteban V, Ruperez M, Egido J, Ruiz-Ortega M . Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism . Circulation 2005 ; 111 : 2509 - 2517
Ruiz-Ortega M, Ruperez M, Esteban V, Egido J . Molecular mechanisms of angiotensin II-induced vascular injury . Curr Hypertens Rep 2003 ; 5 : 73 - 79
Ruiz-Ortega M, Rodríguez-Vita J, Sanchez-Lopez E, Carvajal G, Egido J . TGF-beta signaling in vascular fibrosis . Cardiovasc Res 2007 ; 74 : 196 - 206
Sanyal S, Kim JY, Kim HJ, Takeda J, Lee YK, Moore DD, Choi HS . Differential regulation of the orphan nuclear receptor small heterodimer partner (SHP) gene promoter by orphan nuclear receptor ERR isoforms . J Biol Chem 2002 ; 277 : 1739 - 1748
Seol W, Choi HS, Moore DD . An orphan nuclear hormone receptor that lacks a DNA binding domain and heterodimerizes with other receptors . Science 1996 ; 272 : 1336 - 1339
Seol W, Hanstein B, Brown M, Moore DD . Inhibition of estrogen receptor action by the orphan receptor SHP (short heterodimer partner) . Mol Endocrinol 1998 ; 12 : 1551 - 1557
Sobel BE, Taatjes DJ, Schneider DJ . Intramural plasminogen activator inhibitor type-1 and coronary atherosclerosis . Arterioscler Thromb Vasc Biol 2003 ; 23 : 1979 - 1989
Suh JH, Huang J, Park YY, Seong HA, Kim D, Shong M, Ha H, Lee IK, Lee K, Wang L, Choi HS . Orphan nuclear receptor small heterodimer partner inhibits transforming growth factor-beta signaling by repressing SMAD3 transactivation . J Biol Chem 2006 ; 281 : 39169 - 39178
van Leeuwen RT, Kol A, Andreotti F, Kluft C, Maseri A, Sperti G . Angiotensin II increases plasminogen activator inhibitor type 1 and tissue-type plasminogen activator messenger RNA in cultured rat aortic smooth muscle cells . Circulation 1994 ; 90 : 362 - 368
Vaughan DE, Lazos SA, Tong K . Angiotensin II regulates the expression of plasminogen activator inhibitor-1 in cultured endothelial cells. A potential link between the renin-angiotensin system and thrombosis . J Clin Invest 1995 ; 95 : 995 - 1001
Vaughan DE . PAI-1 and TGF-β: unmasking the real driver of TGF-β-induced vascular pathology . Arterioscler Thromb Vasc Biol 2006 ; 26 : 679 - 680
Wang TJ, Gona P, Larson MG, Tofler GH, Levy D, Newton-Cheh C, Jacques PF, Rifai N, Selhub J, Robins SJ, Benjamin EJ, D'Agostino RB, Vasan RS . Multiple biomarkers for the prediction of first major cardiovascular events and death . N Engl J Med 2006 ; 355 : 2631 - 2639
Wang W, Huang XR, Canlas E, Oka K, Truong LD, Deng C, Bhowmick NA, Ju W, Bottinger EP, Lan HY . Essential role of Smad3 in angiotensin II-induced vascular fibrosis . Circ Res 2006 ; 98 : 1032 - 1039
Weir MR, Dzau VJ . The renin-angiotensin-aldosterone system: a specific target for hypertension management . Am J Hypertens 1999 ; 12 : 205 - 213
Weisberg AD, Albornoz F, Griffin JP, Crandall DL, Elokdah H, Fogo AB, Vaughan DE, Brown NJ . Pharmacological inhibition and genetic deficiency of plasminogen activator inhibitor-1 attenuates angiotensin II/salt-induced aortic remodeling . Arterioscler Thromb Vasc Biol 2005 ; 25 : 365 - 371
Yamagata K, Daitoku H, Shimamoto Y, Matsuzaki H, Hirota K, Ishida J, Fukamizu A . Bile acids regulate gluconeogenic gene expression via small heterodimer partner-mediated repression of hepatocyte nuclear factor 4 and Foxo1 . J Biol Chem 2004 ; 279 : 23158 - 23165
Acknowledgements
We would like to thank Dr. David D. Moore for permission to use SHP KO mice. This work was supported by the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korea government (MEST) (R01-2007-000-10057-0 to K. G. P.; 2009-0063267 to I. K. L.; 2006-2005412 to K. U. L) and the Korea Research Foundation Grant funded by the Korean Government (MOEHRD, Basic Research Promotion Fund) (KRF-2008-331-E00121 to K. G. P.).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lee, KM., Seo, HY., Kim, MK. et al. Orphan nuclear receptor small heterodimer partner inhibits angiotensin II-stimulated PAI-1 expression in vascular smooth muscle cells. Exp Mol Med 42, 21–29 (2010). https://doi.org/10.3858/emm.2010.42.1.002
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2010.42.1.002
