
Abstract
In addition to its well-known glycolytic activity, GAPDH displays multiple functions, such as nuclear RNA export, DNA replication and repair, and apoptotic cell death. This functional diversity depends on its intracellular localization. In this study, we explored the signal transduction pathways involved in the nuclear translocation of GAPDH using confocal laser scanning microscopy of immunostained human diploid fibroblasts (HDFs). GAPDH was present mainly in the cytoplasm when cultured with 10% FBS. Serum depletion by culturing cells in a serum-free medium (SFM) led to a gradual accumulation of GAPDH in the nucleus, and this nuclear accumulation was reversed by the re-addition of serum or growth factors, such as PDGF and lysophosphatidic acid. The nuclear export induced by the re-addition of serum or growth factors was prevented by LY 294002 and SH-5, inhibitors of phosphoinositide 3-kinase (PI3K) and Akt/protein kinase B, respectively, suggesting an involvement of the PI3K signaling pathway in the nuclear export of GAPDH. In addition, 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR), an activator of AMP-activated protein kinase (AMPK), stimulated the nuclear translocation of GAPDH and prevented serum- and growth factor-induced GAPDH export. AMPK inhibition by compound C or AMPK depletion by siRNA treatment partially prevented SFM- and AICAR-induced nuclear translocation of GAPDH. Our data suggest that the nuclear translocation of GAPDH might be regulated by the PI3K signaling pathway acting mainly as a nuclear export signal and the AMPK signaling pathway acting as a nuclear import signal.
Similar content being viewed by others

Introduction
GAPDH (E.C. 1.2.1.12) functions as a glycolytic enzyme working within the cytoplasm. GAPDH forms a tetramer to be active and catalyzes the oxidative phosphorylation of glyceraldehyde 3-phosphate, producing 1,3-bisphosphoglycerate. Cysteine and histidine are located in the active site. Besides its role in glycolysis, GAPDH is also involved in several biological functions, including the organization of the cytoskeleton and regulation of endocytosis, binding and transport of tRNA, and regulation of translation, transcription, replication, DNA repair, cell proliferation, and apoptosis (Sirover, 2005; Harada et al., 2007). Its diverse activities appear to be regulated according to its oligomeric structure and subcellular localization to the nucleus and other subcellular organelles such as mitochondria and the cytoskeleton (Barbini et al., 2007). The nuclear translocation of GAPDH upon apoptosis (Sawa et al., 1997; Shashidharan et al., 1999; Du et al., 2007), serum deprivation (Schmitz, 2001; Schmitz et al., 2003), oxidative stresses (Dastoor and Dreyer, 2001), and genotoxic stresses (Brown et al., 2004) has been reported.
Analysis of the growth factor signaling pathway with specific inhibitors has revealed that the nuclear export of GAPDH is prevented by LY 294002, an inhibitor of phosphoinositide 3-kinase (PI3K) (Schmitz, 2001). Because PI3K links the growth factor signaling pathway with cell death via the repression of an apoptotic inducer, they have suggested that the nuclear accumulation of GAPDH upon growth factor depletion might be reversed by PI3K-induced survival signals (Schmitz, 2001). Activated PI3K produces phosphatidyl inositol 3,4,5-triphosphate (PIP3), which interacts with a variety of molecules, such as Akt kinase (also called PKB) via pleckstrin homology (PH) domains of these downstream targets (Cooray, 2004). The interaction of PIP3 with Akt allows the phosphorylation of Akt by PDK1/2 at two key regulatory sites, Thr308 and Ser473 near the C-terminus, and the subsequent activation of Akt (Rameh and Cantley, 1999). Akt is a serine/threonine kinase that phosphorylates many different target proteins including GSK-3β, Bad, caspase-9, forkhead transcription factors and NF-kB (Cooray, 2004).
AMP-activated protein kinase (AMPK) is a heterotrimeric serine/threonine protein kinase consisting of a catalytic α subunit and regulatory β and γ subunit. AMPK activity can be regulated by a low energy state (increases in the AMP/ATP ratio), phosphorylation of Thr172 and Ser485/491 of the α subunit, and stresses such as reactive oxygen species (ROS), hypoxia, and genotoxic drugs (Stein et al., 2000; Hawley et al., 2003; Hurley et al., 2006). AMPK has various cellular functions, including the regulation of cellular metabolism, ion channels, and gene expression (Hardie, 2004), activation of glucose transport during hypoxia and ischemia via eNOS phosphorylation (Li et al., 2004), cell cycle arrest via p53 phosphorylation (Jones et al., 2005), differentiation of endothelial progenitor cells via eNOS activation (Li et al., 2008), inhibition of protein synthesis via TSC1/2, induction of aortic vasorelaxation in mice (Goirand et al., 2007), and regulation of apoptotic cell death signals (Cao et al., 2008; Kim et al., 2008; Lee et al., 2009).
Since both PI3K/Akt and AMPK are responsive to serum and growth factors, and an AMPK activator, 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR), has been shown to inhibit the PI3K signaling pathway (Jhun et al., 2004), we postulated that the PI3K and AMPK signal transduction pathways might contribute to GAPDH translocation. Following serum depletion (by culturing in serum-free medium, SFM) and the re-addition of serum (by culturing in FBS), PDGF, and lysophosphatidic acid (LPA), we examined the intracellular distribution of GAPDH by confocal laser scanning microscopy of immunostained human diploid fibroblasts (HDFs). The involvement of the PI3K signaling pathway was examined using inhibitors of PI3K and Akt, LY 294002 and SH-5, respectively. The role of AMPK was examined using an inhibitor of AMPK (compound C), an activator of AMPK (AICAR), and siRNAs of AMPK. In addition, we examined the involvement of NOS and p53 in GAPDH nuclear localization using NOS inhibitors, N-nitro-L-arginine methyl ester (L-NAME) and N-monomethyl-L-arginine (L-NMMA), and p53-null cells, MEF-p53-/- and HCT116-p53-/-.
Results
Reversible translocation of GAPDH between the cytosol and nucleus by the availability of serum and growth factors
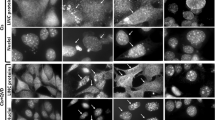
Because serum depletion had no influence on the total GAPDH expression level, we investigated the intracellular distribution of GAPDH by confocal laser scanning microscopy of immunostained HDFs (Figures 1A and 1B). Cells in which green FITC fluorescence was present in the cytosol, but absent or weak in the nuclear fraction, were counted as cells with cytosolic GAPDH. Cells with dense bright green fluorescence only in the nucleus were counted as cells with nuclear GAPDH. Cells containing green fluorescence evenly in both the nuclear and cytosolic fractions were counted as cells with cytosolic and nuclear GAPDH (Figure 1B). To compare the distribution of GAPDH between the cytosol and nucleus, the percent distributions of cells with cytosolic GAPDH (cytosol: black bar) or nuclear GAPDH alone (nuclear: white bar), and cells with both cytosolic and nuclear GAPDH (cytosol + nuclear: gray bar) were plotted in Figure 1C.

Effect of serum depletion on the intracellular distribution of GAPDH. HDFs were maintained in culture medium containing 10% FBS for 2 days and then serum-depleted by incubation with SFM for the times indicated (1-5 days). Cells were immunostained with monoclonal anti-GAPDH antibody (1:1,000) and FITC-conjugated anti-mouse secondary antibodies, and analyzed by confocal laser scanning microscopy. Immunostained cells without (10% FBS) and with serum depletion for 3 days (SFM-3d) or 5 days (SFM-5d) are shown in (A). Representative cell with green FITC fluorescence present in the cytosol, but absent or weak in the nuclear fraction (cytosolic GAPDH), cell with dense, bright green fluorescence only in the nucleus (nuclear GAPDH, cell with green fluorescence equally distributed in both the nuclear and cytosolic fractions (cytosol + nuclear GAPDH) are shown in (B). The number of cells with cytosolic GAPDH alone (cytosol: black bar), nuclear and cytosolic GAPDH (cytosol + nuclear: gray bar) or nuclear GAPDH alone (nuclear: white bar) was counted and the percentage distributions are plotted as means ± standard deviations in (C). The results shown are representative of three independent experiments.
Almost all GAPDH was present in the cytosol of HDFs with a population doubling of less than 25 (designated as young HDFs in other reports) when they were cultured in DMEM containing 10% FBS for 2 days (Figure 1A, 10% FBS and Figure 1C, FBS). Serum withdrawal from young HDFs for 1 day initiated an accumulation of GAPDH in the nucleus, and its nuclear accumulation increased gradually until 5 days (Figure 1A, 10% FBS/SFM-5d). The number of cells with cytosolic GAPDH alone decreased such that the number of cells with nuclear GAPDH increased gradually to over 90% in young cells with serum depletion for 4-5 days (Figure 1C, SFM).
Nuclear export of GAPDH to the cytosol was observed upon the addition of serum to HDFs that had been serum-deprived for 5 days (Figure 2A, SFM-5d/10% FBS). After 2 days of incubation with 10% FBS, over 90% of the GAPDH had been released back to the cytosol (Figures 2A and 2B). Our data suggest that GAPDH nuclear translocation is a reversible process in HDFs. A portion (~60%) of the nuclear GAPDH in serum-deprived cells was also exported by treatment with 100 ng/ml PDGF for 24-48 h (Figure 2C) or 30 µM LPA for 48 h (Figure 2D).

Effect of serum and growth factor re-addition on the nuclear GAPDH export after serum depletion. HDFs grown on 10% FBS culture medium were serum-depleted by incubation in SFM for 5 days. Next, 10% FBS was re-added to the serum-depleted cells for the times indicated (1-3 days). Cells were immunostained against GAPDH and analyzed by confocal laser scanning microscopy, as described in Figure 1. Immunostained cells with serum depletion for 5 days (SFM-5d) and re-addition of 10% FBS for 1-3 days (SFM-5d/10% FBS-1d to -3d) are shown in (A). The number of cells with cytosolic GAPDH alone, nuclear and cytosolic GAPDH, or nuclear GAPDH alone was counted and the percentage distributions are plotted in (B), as described in Figure 1B. Instead of FBS, 100 ng/ml PDGF (C) or 30 µM LPA (D) was re-added to the serum-depleted cells for the times indicated (1-48 h).
PI3K-dependent export of nuclear GAPDH to the cytoplasm
The cell number with nuclear GAPDH alone was combined with that including both nuclear and cytosolic GAPDH to illustrate the effects of inhibitors and activators (nuclear +/- cytosol in Figures 3-5 and 7-9). An inhibitor of PI3K, LY 294002, induced the nuclear accumulation of GAPDH significantly (P < 0.001) at concentrations of 10-50 µM, even when cultured in 10% FBS for 2 days (Figure 3A). After serum depletion for 5 days, the medium was replaced with 10% FBS, 100 ng/ml PDGF, or 30 µM LPA in the presence and absence of 50 µM LY 294002. As shown in Figure 3B, the nuclear export induced by the re-addition of 10% FBS, PDGF, or LPA was (P < 0.001) prevented completely by the PI3K inhibitor, LY 294002.

Effect of inhibitors of PI3K and Akt on the intracellular distribution of GAPDH. (A) HDFs were incubated in culture medium containing 10% FBS and treated with vehicle (DMSO) or various concentrations of the PI3K inhibitor LY 294002 (10-50 µM) for 2 days. Cells were immunostained against GAPDH and analyzed by confocal laser scanning microscopy, as described in Figure 1. The number of cells with cytosolic GAPDH alone (cytosol: black bar) and cells having nuclear GAPDH with or without cytosolic GAPDH (nuclear +/- cytosol: red bar) was counted and the percent distributions are plotted as means ± standard deviations. ***P < 0.001, comparison between the FBS control and FBS + LY 294002 (µM). (B) HDFs were serum-depleted by incubating in SFM for 4 days, and then either 10% FBS, 100 ng/ml PDGF, or 30 µM LPA was re-added to serum-depleted cells in the absence (-) or presence (+) of 50 µM LY 294002 for 2 days. **P < 0.01 and ***P < 0.001, comparison between the absence (-) and presence (+) of LY 294002. (C) HDFs were incubated in medium containing 10% FBS and treated with vehicle (DMSO) or various concentrations of the Akt inhibitor SH-5 (5-20 µM) for 2 days. **P < 0.01, comparison between the FBS control and FBS + SH-5 (µM). (D) HDFs were serum-depleted by incubating in SFM for 4 days, and then either 10% FBS, 100 ng/ml PDGF, or 30 µM LPA was re-added to serum-depleted cells in the absence (-) or presence (+) of 20 µM SH-5 for 2 days. **P < 0.01 and ***P < 0.001, comparison between the absence (-) and presence (+) of SH-5. The results shown are representative of more than three independent experiments. Serum-starved HDF cells were pre-incubated with vehicle (-), 50 µM LY 294002 (E, +), or 20 µM SH-5 (F, +). Cells were treated with 10% FBS/DMEM for 30 min and then lysed. Total cell extract (30 µg per lane) was resolved by 10% PAGE and analyzed by Western blot analysis, using specific antibodies against phospho (Ser473)-Akt, total Akt, phospho (Ser8)-GSK-3β, total GSK-3β, phospho-Bad, total Bad, and β-actin as a control (1:1,000 dilutions).

Effect of the adenosine kinase inhibitor 5'-ITC on the GAPDH distribution. (A) HDFs were incubated in medium containing 10% FBS and treated with vehicle (DMSO) or various concentrations (0.1-2 µM) of the adenosine kinase inhibitor 5'-ITC for 2 days. Cells were immunostained and analyzed by confocal laser scanning microscopy, as described in Figure 1. The number of cells with cytosolic GAPDH alone (cytosol) and cells having nuclear GAPDH with or without cytosolic GAPDH (nuclear +/- cytosol) was counted, and the percent distributions are plotted as means ± standard deviations. ***P < 0.001, comparison between the FBS control and FBS + 5'-ITC (µM). (B) HDFs were serum-depleted by incubating in SFM for 4 days (SFM-4d) and then 10% FBS was re-added to serum-depleted cells and treated with vehicle (0) or various concentrations of 5'-ITC (0.1-2 µM) for 2 days. ***P < 0.001, comparison between the FBS control (0) and FBS + 5'-ITC (µM) after SFM-4d. (C) HDFs were incubated in a culture medium containing 10% FBS for 2 days and then treated with 2 mM AICAR in the absence (0) or presence of various concentrations of 5'-ITC (0.1-2 µM) for 2 days. ***P < 0.001, comparison between 2 mM AICAR only without compound C (0) and 2 mM AICAR + 5'-ITC (µM). (D) HDFs were serum-depleted by incubating in SFM for 4 days (SFM-4d), and then 10% FBS was re-added to serum-depleted cells with 2 mM AICAR in the absence (0) or presence of various concentrations of 5'-ITC (0.1-2 µM) for 2 days. ***P < 0.001, comparison between the AICAR/10%FBS control (0) and AICAR/10%FBS + 5'-ITC (µM) after SFM-4d.

Detection of phosphorylated AMPK and its substrate ACC by Western blot analysis. (A) HDF cells were serum-starved by incubation with SFM for the times indicated (1-4 days) in the absence (-) or presence (+) of 10 µM of the AMPK inhibitor compound C. Cells were lysed and total cell extract (30 µg per lane) was resolved by 10% PAGE and blotted onto Immobilon PVDF membranes for Western blot analysis, as described in the Methods. Monoclonal anti-phospho (Thr172)-AMPK and anti-phospho (Ser79) ACC antibodies were used at 1:1,000 dilution and polyclonal anti-pan-AMPK and anti-pan-ACC antibodies were used at 1:500 dilution. (B) HDF cells were treated with vehicle (-) or AICAR (+) in medium containing 10% FBS for short-term periods (0.5-2 h) in the absence (-) or presence (+) of 10 µM compound C. Primary and secondary antibodies were the same as those used in (A). (C) HDF cells were treated with vehicle (-) or AICAR (+) in medium containing 10% FBS for a long-term period (2 days) in the absence (-) or presence (+) of 10 µM compound C. Primary and secondary antibodies were the same as those used in (A).

Effect of NOS inhibitors on SFM- and AICAR-induced GAPDH translocation. HDFs were cultured in 10% FBS medium, and then treated with SFM for 4 days or 2 mM AICAR in 10% FBS in the absence (-) or presence (+) of 100 µM of L-NMMA (A) or 0.5 mg/ml L-NAME (B) for 2 days. Cells were immunostained against GAPDH and analyzed by confocal laser scanning microscopy as described in Figure 1. The number of cells with cytosolic GAPDH alone (cytosol) and cells having nuclear GAPDH with or without cytosolic GAPDH (nuclear +/- cytosol) was counted, and the percent distributions are plotted as means ± standard deviations. *P < 0.05, comparison between the absence (-) and presence (+) of NOS inhibitors.
SH-5, an inhibitor of PI3K-downstream kinase Akt, also induced the nuclear translocation of GAPDH significantly (P < 0.01) at a concentration of 20 µM when cultured in 10% FBS (Figure 3C). This concentration of SH-5 reduced partially but significantly (P < 0.001) the serum-induced GAPDH export (Figure 3D). Because the effect of SH-5 was less than that of LY 294002, we used Western blot analysis to compare the efficacy of LY 294002 and SH-5 on the phosphorylation of downstream substrates such as Akt, GSK3β, and Bad. As shown in Figure 3E, Ly 294002 completely inhibited Akt phosphorylation but only partially inhibited GSK3β phosphorylation. Similarly, SH-5 only partially inhibited the phosphorylation of GSK3β and Bad, two downstream signaling proteins of Akt. Although the reason why the Akt inhibitor is less effective remains elusive, our data suggest that the PI3K/Akt signaling pathway is involved in the nuclear export of GAPDH in HDFs.
AICAR affects nuclear localization of GAPDH
To examine the involvement of AMPK in GAPDH translocation, we first examined the effect of AICAR, a specific AMPK activator, on GAPDH nuclear localization. HDFs were grown in culture medium containing 10% FBS for 2 days and then grown further in fresh culture medium with 0.5-2 mM AICAR for 1-4 days. AICAR by itself stimulated the nuclear translocation of GAPDH in a dose- and time-dependent manner (Figures 4A and 4B, respectively). About 60% of the cytosolic GAPDH was translocated to the nucleus after 4 days of treatment with 0.5 mM AICAR (P < 0.001) and its translocation increased to over 90% by 2 mM AICAR treatment for 4 days (Figures 4A and 4B). After GAPDH had localized to the nucleus by serum depletion for 4 days, the effect of AICAR on the nuclear export of GAPDH by FBS was also examined. In the presence of 0.5-2 mM AICAR, nuclear GAPDH was not exported to the cytosol upon serum re-addition (Figure 4C).

Effect of AICAR on GAPDH distribution. (A) HDFs were incubated in medium containing 10% FBS and treated with vehicle (PBS) or various concentrations of AICAR (0.5-2 mM) for 4 days. Cells were immunostained and analyzed by confocal laser scanning microscopy, as described in Figure 1. The number of cells with cytosolic GAPDH alone (cytosol) and cells having nuclear GAPDH with or without cytosolic GAPDH (nuclear +/- cytosol) was counted and the percent distributions are plotted as means ± standard deviations. ***P < 0.001, comparison between the FBS control and FBS + AICAR (mM). (B) HDFs were incubated in medium containing 10% FBS and treated with vehicle (PBS) or 2 mM AICAR for the times indicated (1-4 days). ***P < 0.001, comparison between the FBS control and FBS + 2 mM AICAR. (C) HDFs were serum-depleted by incubating in SFM for 4 days (SFM-4d), and then 10% FBS was re-added to serum-depleted cells and treated with vehicle (0) or various concentrations of AICAR (0.5-2 mM) for 2 days. ***P < 0.001, comparison between the FBS control (0) and FBS + AICAR (mM) after SFM-4d. (D) HDFs were serum-depleted by incubating in SFM for 4 days, and then either 10% FBS, 100 ng/ml PDGF, or 30 µM LPA was re-added to serum-depleted cells in the absence (-) or presence (+) of 1 mM AICAR for 2 days. **P < 0.01 and ***P < 0.001, comparison between the absence (-) and presence (+) of 1 mM AICAR. The results shown are representative of more than three independent experiments.
To work as a specific activator of AMPK, AICAR should be internalized and become phosphorylated by adenosine kinase to form AICAR-monophosphate (AICA ribotide, ZMP) (Vincent et al., 1996). ZMP mimics the effects of AMP on AMPK (Corton et al., 1995). Therefore, we first examined whether the AICAR-induced nuclear accumulation of GAPDH depends on AICAR uptake and subsequent phosphorylation to ZMP using an adenosine kinase inhibitor, 5'-iodotubercidin (5'-ITC). Within the concentration range of 0.1-1 µM, 5'-ITC increased a low but significant (P < 0.001) nuclear export of GAPDH in a dose-dependent manner, while at higher concentrations, such as 2 µM, it enhanced the level of nuclear GAPDH slightly (Figure 5A). After serum depletion for 4 days, FBS-induced GAPDH export was also examined in the presence of 5'-ITC. The lower concentrations of 5'-ITC did not affect FBS-induced GAPDH export, but 5'-ITC concentrations as high as 2 µM resulted in about 70% nuclear GAPDH (Figure 5B). The AICAR-induced nuclear accumulation was partially but significantly (P < 0.001) reduced in the presence of various doses of 5'-ITC, compared to the level observed in the presence of 2 mM AICAR only (Figure 5C). Over 90% of the cytosolic GAPDH was translocated to the nucleus after 4 days of treatment with SFM, and then most GAPDH was released by the addition of 10% FBS. We also observed that 2 mM AICAR blocked FBS-induced release, and this AICAR effect was reduced significantly (P < 0.001) in the presence of lower concentrations (0.1-0.5 µM) of 5'-ITC (Figure 5D). Our data indicate that the AMPK activation via ZMP production from AICAR may contribute to the AICAR-induced nuclear GAPDH accumulation. Because the nuclear GAPDH increased at the higher concentrations (1-2 µM) of 5'-ITC under most conditions, these 5'-ITC concentrations may exert a cellular toxic effect, resulting in nuclear GAPDH accumulation.
AMPK is involved in SFM- and AICAR-dependent nuclear translocation of GAPDH
To confirm that SFM- and AICAR-induced GAPDH nuclear translocation was mediated through AMPK activation, we examined the effect of an AMPK inhibitor, compound C, on the GAPDH distribution. Compound C in the culture medium with 10% FBS induced GAPDH translocation to the nucleus significantly (P < 0.001) in a dose- and time-dependent manner (Figures 6A and 6B, respectively). Moreover, compound C prevented SFM- and AICAR-induced GAPDH translocation (Figures 6C and 6D, respectively).

Effect of the AMPK inhibitor compound C on the GAPDH distribution. (A) HDFs were incubated in medium containing 10% FBS and treated with vehicle (DMSO) or various concentrations of the AMPK inhibitor compound C (1-10 µM) for 2 days. Cells were immunostained and analyzed by confocal laser scanning microscopy, as described in Figure 1. The number of cells with cytosolic GAPDH alone (cytosol) and cells having nuclear GAPDH with or without cytosolic GAPDH (nuclear +/- cytosol) was counted and the percent distributions are plotted as means ± standard deviations. ***P < 0.001, comparison between the FBS control and FBS + compound C (µM). (B) HDFs were incubated in medium containing 10% FBS and treated with vehicle (DMSO) or 10 µM of compound C for the times indicated (1-4 days). **P < 0.01 and ***P < 0.001, comparison between the FBS control and FBS + 10 µM compound C. (C) HDFs grown in 10% FBS culture medium were serum-depleted by incubating in SFM for 4 days (SFM-4d) without (0) or with various concentrations of compound C (1-10 µM). ***P < 0.001, comparison between SFM-4d without compound C (0) and SFM-4d + compound C (µM). (D) HDFs were incubated in medium containing 10% FBS for 2 days and then treated with vehicle (PBS) or 2 mM AICAR in the absence (0) or presence of various concentrations of compound C (1-10 µM) for 2 days. ***P < 0.001, comparsion between 2 mM AICAR only without compound C (0) and 2 mM AICAR + compound C (µM). The results shown are representative of more than three independent experiments.
Because the inhibition of SFM- and AICAR-induced GAPDH translocation by 10 µM compound C was significant (P < 0.001) but only partial (about 60%), we examined the effect of compound C on SFM- and AICAR-induced AMPK activation by measuring the phosphorylation status of AMPK and its target substrate (acetyl-CoA carboxylase: ACC) through Western blot analysis. A partial inhibitory effect of 10 µM compound C on SFM-induced phosphorylation of AMPK and ACC was observed (Figure 7A). In addition, the same amount of compound C partially prevented the AICAR-induced phosphorylation of AMPK and ACC during a short-term (0.5-2 h), as well as a long-term incubation period (2 days) (Figures 7B and 7C, respectively). These findings indicate that the partial inhibitory effect of compound C might be due to a partial inhibition of AMPK activity.
We also applied an siRNA approach, specific for AMPK, to abolish the expression of AMPK proteins. AMPKα1 and AMPKα2 are the major isoforms of the AMPKα catalytic subunit in HDFs. Therefore, we utilized a mixture of siRNAs to block the expressions of both AMPKα1 and AMPKα2. As depicted in Figure 8A, treatment with AMPKα1/α2 siRNAs for 2 days reduced the total AMPK protein expression and AMPK phosphorylation induced by SFM and AICAR. AMPK depletion by siRNA treatment significantly (P < 0.001) prevented SFM- and AICAR-induced nuclear translocation of GAPDH compared to the mock control group (Figures 8B and 8C, respectively). Our data indicate that AMPK is involved in SFM- and AICAR-induced GAPDH translocation in HDFs.

SFM- and AICAR-induced GAPDH translocation in AMPK siRNA-transfected HDFs. HDFs were grown in 10% FBS medium for 1 day and transfected with siRNAs of AMPKα1/α2 and a siCONTROL complete kit as a negative mock control for 2 days. Cells were then serum-depleted by incubation with SFM or treated with 2 mM AICAR for 2 days. During the serum depletion or AICAR treatment period, either the mock control siRNAs or AMPKα1/α2 siRNAs were included. (A) Cells were harvested 4 days after transfection, and lysates containing the same amount of protein (45 µg) were analyzed by Western blot using monoclonal anti-phospho-Thr172-AMPK antibody and polyclonal anti-AMPKα1/2 and anti-β-actin antibodies. (B) HDF cells were transfected with siRNAs and treated with SFM (SFM-2d), as described in (A). Cells were immunostained against GAPDH and analyzed by confocal laser scanning microscopy, as described in Figure 1. The number of cells with cytosolic GAPDH alone (cytosol) and cells having nuclear GAPDH with or without cytosolic GAPDH (nuclear +/- cytosol) was counted, and the percent distributions are plotted as means ± standard deviations. ***P < 0.001, comparison between the mock control and AMPK-siRNA. (C) HDF cells were transfected with siRNAs and treated with 2 mM AICAR, as described in (A). Cells were immunostained against GAPDH and analyzed by confocal laser scanning microscopy. ***P < 0.001, comparison between the mock control and AMPK-siRNA.
NOS- and p53-independent nuclear localization of GAPDH
Previous studies have suggested that S-nitrosylation of GAPDH is required for its nuclear translocation in apoptotic cancer cells (Hara et al., 2005; Hara and Snyder, 2006; Du et al., 2007). However, whether S-nitrosylation is necessary for GAPDH nuclear translocation in all cell types with various stimuli is not known. Therefore, the involvement of S-nitrosylation in SFM- and AICAR-induced GAPDH nuclear localization in HDF cells was examined using the NOS inhibitors L-NMMA and L-NAME. We found that GAPDH translocation by SFM and AICAR in cells treated with NOS inhibitors was similar to that observed in the untreated control cells (Figures 9A and 9B, respectively), indicating that NOS activation is not necessary for SFM- and AICAR-induced GAPDH translocation.
p53 has been demonstrated to be phosphorylated and activated by AMPK (Jones et al., 2005). The treatment of cells with AICAR caused cell cycle arrest in S-phase accompanied with increased expression of p21, p27, and p53 proteins (Rattan et al., 2005). In addition, p53 could be activated by GAPDH nuclear translocation via the formation of a GAPDH-Siah1 complex and subsequent activation of p300/CBP in the presence of apoptotic stimuli (Sen et al., 2008). To determine whether p53 might contribute to GAPDH localization, we performed experiments using p53-null (p53-/-) MEF and HCT116 cancer cells. SFM and AICAR could induce GAPDH nuclear accumulation in both normal and p53-null MEF cells (Figures 10A and 10B, respectively). The effects of SFM and AICAR on GAPDH nuclear accumulation were significantly (P < 0.001) higher in p53-null MEF cells than in normal MEF cells. However, p53-null HCT116 cells exhibited little reduction (P < 0.05 at day 2, P < 0.001 at day 4) in SFM-induced GAPDH nuclear localization compared to normal HCT116 cells (Figure 10C). We observed no changes in AICAR-induced GAPDH nuclear localization between normal and p53-null HCT116 cells (Figure 10D). Our data suggest that p53 activation may not be necessary for SFM- and AICAR-induced GAPDH translocation in both MEF cells and HCT116 cancer cells. The activation of p53 might play an inhibitory role in GAPDH nuclear localization at least in the MEF cell system.

SFM- and AICAR-induced GAPDH translocation in p53-null cells. Normal and p53-null MEF cells (A and B) and HCT116 cells (C and D) were cultured in 10% FBS medium and then treated with SFM for the times indicated (A and C) or with various concentrations of AICAR in 10% FBS for 2 days (B and D). Cells were immunostained against GAPDH and analyzed by confocal laser scanning microscopy, as described in Figure 1. The number of cells with cytosolic GAPDH alone (cytosol) and cells having nuclear GAPDH with or without cytosolic GAPDH (nuclear +/- cytosol) was counted, and the percent distributions are plotted as means ± standard deviations. *P < 0.05 and ***P < 0.001, comparison between normal cells and p53-null cells.
Leptomycin B stimulates nuclear accumulation of GAPDH by inhibiting GAPDH export
Recently, the CRM1(chromosome region maintenance 1, also called exportin1/Xpo1)-dependent nuclear export system has been demonstrated to operate in determining GAPDH localization (Brown et al., 2004). Because the CRM1-dependent nuclear export can be inhibited by leptomycin B (LMB), we examined the effect of different LMB concentrations (1-20 ng/ml) in the culture medium with 10% FBS for 2 days (Figure 11A) as well as the effect of 10 ng/ml LMB treatment for 1-4 days (Figure 11B). LMB induced the nuclear accumulation of GAPDH at a dose as low as 1 ng/ml within 1 day. The nuclear fraction increased to about 50% after 2 days of treatment with 10 ng/ml LMB. The LMB-induced nuclear accumulation of GAPDH reached a plateau at 50%, even after longer treatment periods or in the presence of higher concentrations of LMB. We also tested the effect of LMB on GAPDH export by the re-addition of 10% FBS for 2 days following complete serum depletion for 4 days. LMB completely prevented FBS-induced GAPDH export at a dose as low as 1 ng/ml (Figure 11C). These data suggest that GAPDH translocation by serum and growth factors can be regulated via the CRM1-dependent nuclear export system.

Effect of the CRM1 inhibitor LMB on the GAPDH distribution. HDFs were cultured in 10% FBS medium for 2 days and treated with vehicle (methanol) or various concentrations of LMB (1-20 ng/ml) for 2 days (A) or with 10 ng/ml LMB for the times indicated (B). In another experiment (C), HDFs were cultured in 10% FBS medium and serum-depleted by incubation with SFM for 4 days (SFM-4d). FBS was added to serum-depleted cells in the absence (0) or presence of various concentrations of LMB for 2 days. Cells were immunostained against GAPDH and analyzed by confocal laser scanning microscopy, as described in Figure 1. The number of cells with cytosolic GAPDH alone (cytosol) and cells having nuclear GAPDH with or without cytosolic GAPDH (nuclear +/- cytosol) was counted, and the percent distributions are plotted as means ± standard deviations. ***P < 0.001, comparison between the FBS control (0) and FBS + LMB (ng/ml).
Discussion
GAPDH contains a novel CRM1-dependent nuclear export signal (NES) comprising 13 amino acids (KKVVKQASEGPLK) in the C-terminal domain, and truncation or mutation of this NES abrogated the binding of CRM1 and caused the nuclear accumulation of GAPDH (Brown et al., 2004). Because treatment of cells with the inhibitor LMB did not influence the cytoplasmic localization of GFP-GAPDH in NIH3T3 fibroblasts (Schmitz et al., 2003), the involvement of CRM1 in GAPDH export remains controversial. In our experiment, we found that LMB completely prevented FBS-induced GAPDH export after serum depletion for 4 days. This finding supports a positive role of CRM1 in serum- and growth factor-induced GAPDH nuclear export in HDF cells.
It has been suggested that the nuclear export of estradiol receptor alpha (ERα) depends on CRM1 as well as PI3K/Akt activation (Lombardi et al., 2008). Akt-dependent phosphorylation of a forkhead protein Foxo1 (FKHR) drives its association with ERα, thereby triggering the complex export from the nucleus. In this study, we found that a PI3K inhibitor, LY 29004, inhibited serum- or growth factor-induced GAPDH nuclear export in HDF cells, which is in agreement with previous findings (Schmitz, 2001). Therefore, we suggested PI3K as a candidate that regulates GAPDH nuclear export in HDF cells but its regulation mechanism is not yet clear. A similar forkhead family protein might also be associated with GAPDH, and perhaps Akt-dependent phosphorylation of these forkhead proteins could play a role in GAPDH export. This should be verified further.
GAPDH has been reported to undergo autophosphorylation (Kawamoto and Caswell, 1986) as well as phosphorylation via phosphatidylserine-dependent PKCs (Reiss et al., 1996). Although GAPDH export was not altered upon inhibition of PKC by a general PKC inhibitor bisindolylmaleimide II in NIH3T3 cells (Schmitz, 2001), we could not exclude the possibility that the atypical PKC family PKCι/λ could be involved in GAPDH nuclear translocation. The atypical PKCs are activated by the lipid second messenger PIP3 produced by PI3K. PI3K could be activated by EGF- or PDGF-receptor tyrosine kinases. In our study, we observed that a PI3K inhibitor, LY 294002, provided a dramatic inhibition of GAPDH nuclear export. Moreover, GAPDH has been shown to be phosphorylated by PKCι/λ and influence microtubule dynamics in the early secretory pathway (Tisdale, 2002, 2003; Tisdale et al., 2004). Src-dependent PKCι/λ tyrosine phosphorylation is required for PKCι/λ association with Rab2 and GAPDH on pre-Golgi intermediates (Tisdale and Artalejo, 2006). Therefore, it is possible that PIP3-induced atypical PKCι/λ might be involved in GAPDH nuclear translocation.
Because GAPDH also possesses a region homologous to a nuclear localization sequence motif (NLS) between amino acids 259 and 263 (KKVVK), its nuclear transport could occur via this NLS (Sirover, 1999). However, possible regulatory proteins and signaling pathways have not been elucidated. In the current study, we found that a well-known AMPK activator AICAR induced GAPDH nuclear translocation and an adenosine kinase inhibitor 5'-ITC abrogated this change. In the presence of AMPK inhibitors and siRNAs for the inhibition of AMPK activity and blockage of AMPK expression, respectively, we also observed an inhibition of GAPDH translocation. These data suggest that AMPK activation via ZMP production from AICAR may play a role in the nuclear accumulation of GAPDH. Because AICAR inhibits the PI3K/Akt signaling pathway in an AMPK-dependent (Peairs et al., 2009) or AMPK-independent manner (Jhun et al., 2004), a part of AICAR effect on GAPDH nuclear accumulation might be explained by the negative effect on the PI3K/Akt signaling pathway.
AMPK directly phosphorylates and activates p53 (Jones et al., 2005) and AICAR increases p53 expression (Rattan et al., 2005). In addition, p53 could be activated by GAPDH nuclear translocation via the formation of GAPDH-Siah1 complexes (Sen et al., 2008). In our experiments, SFM and AICAR induced GAPDH nuclear accumulation in both normal (p53+/+) and p53-null (p53-/-) MEF and HCT116 cells. Therefore, we suggest that p53 activation may not be necessary for SFM- and AICAR-induced GAPDH translocation in MEF and HCT116 cells.
Because AMPK phosphorylates endothelial NOS on Ser633 and control NO bioavailability in vascular endothelial cells (ECs) (Chen et al., 2009), and S-nitrosylated GAPDH can be translocated to the nucleus under diverse apoptotic stimuli (Hara et al., 2005; Hara and Snyder, 2006; Du et al., 2007), we investigated the involvement of NOS in SFM- and AICAR-induced GAPDH nuclear localization using NOS inhibitors. We observed no effects of NOS inhibitors, such as L-NMMA and L-NAME, on the nuclear translocation of GAPDH, suggesting that S-nitrosylation of GAPDH may not be necessary for SFM- and AICAR-induced nuclear translocation in this cell system.
Based on the current findings, we propose signal transduction pathways involved in the nuclear translocation of GAPDH (Figure 12). GAPDH exists mainly in the cytoplasm in the presence of serum and growth factors. When cells are serum-depleted by SFM or treated with AICAR, GAPDH moves to the nucleus. The re-addition of serum or growth factors to serum-depleted cells causes an export of nuclear GAPDH, which requires the PI3K/Akt signal transduction pathway. The inhibition of PI3K and Akt by LY 294002 and SH-5, respectively, prevents GAPDH export and causes GAPDH accumulation in the nucleus. In contrast, the depletion of serum or growth factors, or treatment with AICAR results in AMPK activation, which may play a role in the nuclear translocation of GAPDH.

Proposed signal transduction pathways involved in the nuclear translocation of GAPDH. GAPDH (G in the figure) is present mainly in the cytoplasm in the presence of serum and growth factors. When cells are serum-depleted by incubation with SFM or treated with the AMPK activator AICAR, GAPDH moves to the nucleus. The re-addition of serum or growth factors to serum-depleted cells causes an export of nuclear GAPDH. An export of nuclear GAPDH requires the PI3K/Akt signal transduction pathway. Inhibition of PI3K and Akt by LY 294002 and SH-5, respectively, prevents GAPDH export and causes GAPDH accumulation in the nucleus. GAPDH translocation by serum and growth factors may be regulated via the CRM1-dependent nuclear export system, as judged by the inhibitory effect of LMB on GAPDH export. In contrast, the depletion of serum or growth factors (SFM) results in AMPK activation, which may play a role in the nuclear translocation of GAPDH. AMPK activation by AICAR via ZMP production also causes GAPDH translocation to the nucleus. AMPK inhibition by compound C or AMPK depletion by siRNA treatment prevents SFM- and AICAR-induced nuclear translocation of GAPDH. The inhibition of ZMP production from AICAR by the adenosine kinase inhibitor 5'-ITC reduces the AICAR-induced nuclear accumulation of GAPDH.
Taken together, we conclude that serum-dependent signaling pathways may regulate any import or export processes of GAPDH. The PI3K/Akt signaling pathway regulates the GAPDH nuclear export signal positively or the nuclear import signal negatively. The AMPK signaling pathway, however, works in the opposite way, regulating the GAPDH nuclear import signal positively and its export signal negatively. The involvement of other signaling pathways should be investigated further to explain the molecular mechanism of reversible GAPDH nuclear translocation.
Methods
Materials
The followings are the reagents used in this study and their sources: PDGF-BB, LPA, LMB, FITC-conjugated goat antimouse secondary antibody, and mouse anti-β-actin monoclonal antibody from Sigma-Aldrich (St. Louis, MO); DMEM, FBS, penicillin, and streptomycin from Gibco/BRL Life Technologies, Inc (Carlsbad, CA); PI3K inhibitor LY 294002, anti-rabbit monoclonal antibodies against phospho-AMPKα (Thr172) and GSK-3β (Ser9), anti-mouse monoclonal antibodies against phospho-Bad (Ser136) and phospho-Akt (Ser473), and anti-rabbit polyclonal antibodies against AMPK-α, Akt, and GSK-3β from Cell Signaling Technology (Denver, MA); compound C, AICAR, SH-5, L-NAME, and L-NMMA from Calbiochem (San Diego, CA); mouse anti-GAPDH monoclonal antibody from Chemicon (Bedford, MA); anti-rabbit monoclonal antibodies against ACC1 and phospho-ACC1 (Ser79) from Upstate (Waltham, MA); horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse secondary antibodies from Vector Laboratories (Burlingame, CA); Lipofectamine™ RNAiMAX and mounting solution including DAPI from Invitrogen Life Technologies (Carlsbad, CA); protein assay kit from Bio-Rad Laboratories (Hercules, CA); enhanced chemiluminescence (ECL) system and Amersham hyperfilm™ ECL from GE Healthcare (Buckinghamshire, UK); sense and complement strands of human AMPK-α1 and α2 siRNAs, siCONTROL complete kit, and 5 × siRNA buffer from Dharmacon Inc. (Lafayette, CO); 5'-ITC from Biomol Research Labs (Plymouth Meeting, PA).
Cell culture
Foreskin human fibroblasts were isolated from newborn foreskins as described previously (Boyce and Ham, 1983). Fibroblasts were maintained in 100-mm tissue culture dishes in DMEM supplemented with 10% FBS and 100 U/ml penicillin and 100 µg/ml streptomycin, and maintained in 5% CO2 in a humidified incubator at 37℃. Cells from the early stage of culture (population doubling of less than 25) were utilized for this experiment. Prior to serum withdrawal, cells were grown for 2 days to 60-70% sub-confluence in the culture medium with 10% FBS in DMEM, and then serum-depleted by incubation with serum-free DMEM containing 0.1% bovine serum albumin (SFM) for 1-5 days. To examine the effect of serum and growth factors on GAPDH export from the nucleus, cells were cultured for 2 days in 10% FBS in DMEM, then serum-starved for 4-5 days, and then 10% FBS or agonists (PDGF or LPA) were added for the times indicated. For immunofluorescence staining, cells were cultured on coverslips in 6-well culture plates.
Western blot analysis
Protein expression levels were examined by Western blot analysis, as described previously (Yeo et al., 2000). Total cell lysates were prepared in a lysis buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 2 mM EDTA, 1 mM EGTA, 1 mM Na3VO4, 10 mM NaF, 1 mM DTT, 1 mM PMSF, 25 µg/ml leupeptin, 25 µg/ml aprotinin, 5 mM benzamidine, and 1% Ingepal CA630. Protein concentrations of the lysates were determined using a Bio-Rad protein assay kit, as described by the manufacturer. Cell lysates containing equal amounts of protein were resolved by SDS-PAGE and transferred onto Immobilon PVDF membranes. Blots were blocked with a solution containing 5% nonfat dried milk or 5% BSA and 0.1% Tween 20, and treated with antibodies in the blocking solution overnight, then washed and further probed with HRP-conjugated anti-rabbit IgG (1:5,000). The immune complexes were visualized using an ECL detection system, as described by the manufacturer.
Transfection of siRNAs of human AMPK-α1 and α2
Target sequences for preparing siRNAs of human AMPK-α1 and α2 were as follows: 5'-AGUGAAGGUUGGCAAACAUTT-3' (sense strand) and 5'-AUGUUUGCCAACCUUCACUTT-3' (complement strand) for human AMPK-α1; 5'-GGAAGGUAGUGAAUGCAUATT-3' (sense strand) and 5'-UAUGCAUUCACUACCUUCCTT-3' (complement strand) for human AMPK-α2. A siCONTROL complete kit was used as a negative mock control. Chemically synthesized RNA oligonucleotides were 2'-deprotected, annealed, and desalted as recommended by the manufacturer. RNA duplexes (100 µM stock) resuspended in 1 × siRNA buffer [6 mM HEPES-KOH (pH 7.5), 20 mM KCl, 0.2 mM MgCl2] were transfected with 2.0 × 104 HDF cells at 30-50% confluence. Transfection with siRNAs was performed using Lipofectamine™ RNAiMAX in a 6-well plate as described by the manufacturer. For this purpose, 120 pmol of siRNA duplex was diluted in 100 µl of Opti-MEM I reduced serum medium, and 2 µl of Lipofectamine™ RNAiMAX reagent in 100 µl of Opti-MEM I reduced serum medium, and the diluted siRNA mix and lipid were combined, mixed gently, and incubated for 20 min at room temperature (RT). The lipid and siRNA mix was added to each well (total volume 1.2 ml, 100 nM siRNA). The medium was replaced with culture medium at 4 h after transfection and the cells were incubated for 24-72 h at 37℃. Cells were then analyzed for changes in AMPK protein expression by Western blotting.
Immunofluorescence staining
The cells on coverslips in 6-well culture plates were washed twice with ice-cold PBS and fixed in 4% paraformaldehyde in PBS for 10 min, and then washed once with PBS. Nonspecific protein-binding sites were saturated with blocking solution (2% BSA in TBST: 20 mM Tris, 138 mM NaCl, 0.1% Tween 20, pH 7.4) for 1 h with gentle shaking. The cells were incubated with primary monoclonal antibody against GAPDH (1:1,000 in blocking solution) for 1 h at RT and then washed three times with ice-cold TBST for 10 min each. Cells were incubated with FITC-conjugated goat anti-mouse secondary antibody (1:500) in blocking buffer for 1 h. Cells were washed three times with TBST for 10 min each and dehydrated in three washes of 100% methanol over 1 h. The coverslips were mounted on glass slides with solution including DAPI, which fluorescently (blue) labeled the nuclei. Fluorescence images were then observed by laser scanning confocal microscopy (FV500; Olympus, Tokyo, Japan).
To compare the distribution of GAPDH, both FITC fluorescence (green indicates GAPDH location) and DAPI fluorescence (blue indicates the nuclear region) were examined. After cells were stained for cytosolic GAPDH, nuclear GAPDH, or both cytosolic and nuclear GAPDH, 10-20 different fields of each sample were counted under the microscope at 100 × magnification. The data were combined to give total cell numbers of about 1,000 and were converted to percentage distribution. We repeated this experiment at least three times and statistically analyzed the data.
Statistical analysis
The Student's t-test for paired variables (Office Excel 2007; Microsoft, Redmond, WA) was used to determine whether the index was significantly different as a result of the treatment. Data are presented as the mean ± standard deviation of at least three experiments. P values of less than 0.05 were considered to be statistically significant.
Abbreviations
- AICAR:
-
5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside
- AMPK:
-
AMP-activated protein kinase
- LPA:
-
lysophosphatidic acid
- PI3K:
-
phosphoinositide-3 kinase
- SFM:
-
serum free medium
References
Barbini L, Rodriguez J, Dominguez F, Vega F . Glyceraldehyde-3-phosphate dehydrogenase exerts different biologic activities in apoptotic and proliferating hepatocytes according to its subcellular localization . Mol Cell Biochem 2007 ; 300 : 19 - 28
Boyce ST, Ham RG . Calcium-regulated differentiation of normal human epidermal keratinocytes in chemically defined clonal culture and serum-free serial culture . J Invest Dermatol 1983 ; 81 : 33s - 40s
Brown VM, Krynetski EY, Krynetskaia NF, Grieger D, Mukatira ST, Murti KG, Slaughter CA, Park HW, Evans WE . A novel CRM1-mediated nuclear export signal governs nuclear accumulation of glyceraldehyde-3-phosphate dehydrogenase following genotoxic stress . J Biol Chem 2004 ; 279 : 5984 - 5992
Cao C, Lu S, Kivlin R, Wallin B, Card E, Bagdasarian A, Tamakloe T, Chu WM, Guan KL, Wan Y . AMP-activated protein kinase contributes to UV- and H2O2-induced apoptosis in human skin keratinocytes . J Biol Chem 2008 ; 283 : 28897 - 28908
Chen Z, Peng IC, Sun W, Su MI, Hsu PH, Fu Y, Zhu Y, DeFea K, Pan S, Tsai MD, Shyy JY . AMP-activated protein kinase functionally phosphorylates endothelial nitric oxide synthase Ser633 . Circ Res 2009 ; 104 : 496 - 505
Cooray S . The pivotal role of phosphatidylinositol 3-kinase-Akt signal transduction in virus survival . J Gen Virol 2004 ; 85 : 1065 - 1076
Corton JM, Gillespie JG, Hawley SA, Hardie DG . 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells ? Eur J Biochem 1995 ; 229 : 558 - 565
Dastoor Z, Dreyer JL . Potential role of nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase in apoptosis and oxidative stress . J Cell Sci 2001 ; 114 : 1643 - 1653
Du ZX, Wang HQ, Zhang HY, Gao DX . Involvement of glyceraldehyde-3-phosphate dehydrogenase in tumor necrosis factor-related apoptosis-inducing ligand-mediateddeath of thyroid cancer cells . Endocrinology 2007 ; 148 : 4352 - 4361
Goirand F, Solar M, Athea Y, Viollet B, Mateo P, Fortin D, Leclerc J, Hoerter J, Ventura-Clapier R, Garnier A . Activation of AMP kinase alpha1 subunit induces aortic vasorelaxation in mice . J Physiol 2007 ; 581 : 1163 - 1171
Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, Ferris CD, Hayward SD, Snyder SH, Sawa A . S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding . Nat Cell Biol 2005 ; 7 : 665 - 674
Hara MR, Snyder SH . Nitric oxide-GAPDH-Siah: a novel cell death cascade . Cell Mol Neurobiol 2006 ; 26 : 527 - 538
Harada N, Yasunaga R, Higashimura Y, Yamaji R, Fujimoto K, Moss J, Inui H, Nakano Y . Glyceraldehyde-3-phosphate dehydrogenase enhances transcriptional activity of androgen receptor in prostate cancer cells . J Biol Chem 2007 ; 282 : 22651 - 22661
Hardie DG . The AMP-activated protein kinase pathway--new players upstream and downstream . J Cell Sci 2004 ; 117 : 5479 - 5487
Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG . Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade . J Biol 2003 ; 2 : 28 -
Hurley RL, Barre LK, Wood SD, Anderson KA, Kemp BE, Means AR, Witters LA . Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP . J Biol Chem 2006 ; 281 : 36662 - 36672
Jhun BS, Jin Q, Oh YT, Kim SS, Kong Y, Cho YH, Ha J, Baik HH, Kang I . 5-Aminoimidazole-4-carboxamide riboside suppresses lipopolysaccharide-induced TNF-alpha production through inhibition of phosphatidylinositol 3-kinase/Akt activation in RAW 264.7 murine macrophages . Biochem Biophys Res Commun 2004 ; 318 : 372 - 380
Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB . AMP-activated protein kinase induces a p53-dependent metabolic checkpoint . Mol Cell 2005 ; 18 : 283 - 293
Kawamoto RM, Caswell AH . Autophosphorylation of glyceraldehydephosphate dehydrogenase and phosphorylation of protein from skeletal muscle microsomes . Biochemistry 1986 ; 25 : 657 - 661
Kim HS, Hwang JT, Yun H, Chi SG, Lee SJ, Kang I, Yoon KS, Choe WJ, Kim SS, Ha J . Inhibition of AMP-activated protein kinase sensitizes cancer cells to cisplatin-induced apoptosis via hyper-induction of p53 . J Biol Chem 2008 ; 283 : 3731 - 3742
Lee YK, Park SY, Kim YM, Lee WS, Park OJ . AMP kinase/cyclooxygenase-2 pathway regulates proliferation and apoptosis of cancer cells treated with quercetin . Exp Mol Med 2009 ; 41 : 201 - 207
Li J, Hu X, Selvakumar P, Russell RR, Cushman SW, Holman GD, Young LH . Role of the nitric oxide pathway in AMPK-mediated glucose uptake and GLUT4 translocation in heart muscle . Am J Physiol Endocrinol Metab 2004 ; 287 : E834 - E841
Li X, Han Y, Pang W, Li C, Xie X, Shyy JY, Zhu Y . AMP-activated protein kinase promotes the differentiation of endothelial progenitor cells . Arterioscler Thromb Vasc Biol 2008 ; 28 : 1789 - 1795
Lombardi M, Castoria G, Migliaccio A, Barone MV, Di Stasio R, Ciociola A, Bottero D, Yamaguchi H, Appella E, Auricchio F . Hormone-dependent nuclear export of estradiol receptor and DNA synthesis in breast cancer cells . J Cell Biol 2008 ; 182 : 327 - 340
Peairs A, Radjavi A, Davis S, Li L, Ahmed A, Giri S, Reilly CM . Activation of AMPK inhibits inflammation in MRL/lpr mouse mesangial cells . Clin Exp Immunol 2009 ; 156 : 542 - 551
Rameh LE, Cantley LC . The role of phosphoinositide 3-kinase lipid products in cell function . J Biol Chem 1999 ; 274 : 8347 - 8350
Rattan R, Giri S, Singh AK, Singh I . 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase . J Biol Chem 2005 ; 280 : 39582 - 39593
Reiss N, Hermon J, Oplatka A, Naor Z . Interaction of purified protein kinase C with key proteins of energy metabolism and cellular motility . Biochem Mol Biol Int 1996 ; 38 : 711 - 719
Sawa A, Khan AA, Hester LD, Snyder SH . Glyceraldehyde-3-phosphate dehydrogenase: nuclear translocation participates in neuronal and nonneuronal cell death . Proc Natl Acad Sci USA 1997 ; 94 : 11669 - 11674
Schmitz HD . Reversible nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase upon serum depletion . Eur J Cell Biol 2001 ; 80 : 419 - 427
Schmitz HD, Dutine C, Bereiter-Hahn J . Exportin 1-independent nuclear export of GAPDH . Cell Biol Int 2003 ; 27 : 511 - 517
Sen N, Hara MR, Kornberg MD, Cascio MB, Bae BI, Shahani N, Thomas B, Dawson TM, Dawson VL, Snyder SH, Sawa A . Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis . Nat Cell Biol 2008 ; 10 : 866 - 873
Shashidharan P, Chalmers-Redman RM, Carlile GW, Rodic V, Gurvich N, Yuen T, Tatton WG, Sealfon SC . Nuclear translocation of GAPDH-GFP fusion protein during apoptosis . Neuroreport 1999 ; 10 : 1149 - 1153
Sirover MA . New insights into an old protein: the functional diversity of mammalian glyceraldehyde-3-phosphate dehydrogenase . Biochim Biophys Acta 1999 ; 1432 : 159 - 184
Sirover MA . New nuclear functions of the glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in mammalian cells . J Cell Biochem 2005 ; 95 : 45 - 52
Stein SC, Woods A, Jones NA, Davison MD, Carling D . The regulation of AMP-activated protein kinase by phosphorylation . Biochem J 2000 ; 345 : 437 - 443
Tisdale EJ . Glyceraldehyde-3-phosphate dehydrogenase is phosphorylated by protein kinase Ciota /lambda and plays a role in microtubule dynamics in the early secretory pathway . J Biol Chem 2002 ; 277 : 3334 - 3341
Tisdale EJ . Rab2 interacts directly with atypical protein kinase C (aPKC) iota/lambda and inhibits aPKCiota/lambda-dependent glyceraldehyde-3-phosphate dehydrogenase phosphorylation . J Biol Chem 2003 ; 278 : 52524 - 52530
Tisdale EJ, Artalejo CR . Src-dependent aprotein kinase C iota/lambda (aPKCiota/lambda) tyrosine phosphorylation is required for aPKCiota/lambda association with Rab2 and glyceraldehyde-3-phosphate dehydrogenase on pre-golgi intermediates . J Biol Chem 2006 ; 281 : 8436 - 8442
Tisdale EJ, Kelly C, Artalejo CR . Glyceraldehyde-3-phosphate dehydrogenase interacts with Rab2 and plays an essential role in endoplasmic reticulum to Golgi transport exclusive of its glycolytic activity . J Biol Chem 2004 ; 279 : 54046 - 54052
Vincent MF, Bontemps F, Van den Berghe G . Substrate cycling between 5-amino-4-imidazolecarboxamide riboside and its monophosphate in isolated rat hepatocytes . Biochem Pharmacol 1996 ; 52 : 999 - 1006
Yeo EJ, Hwang YC, Kang CM, Choy HE, Park SC . Reduction of UV-induced cell death in the human senescent fibroblasts . Mol Cells 2000 ; 10 : 415 - 422
Acknowledgements
This work was supported by grants from the National Research Foundation of Korea (NRF) through the Ageing and Apoptosis Research Center at Seoul National University (RII-2002-097-08001-0 and RII-2002-097-08003-0) and from the Korea Basic Science Institute (K-MeP).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kwon, H., Rhim, J., Jang, IS. et al. Activation of AMP-activated protein kinase stimulates the nuclear localization of glyceraldehyde 3-phosphate dehydrogenase in human diploid fibroblasts. Exp Mol Med 42, 254–269 (2010). https://doi.org/10.3858/emm.2010.42.4.025
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2010.42.4.025
Keywords
This article is cited by
-
Shotgun proteomics of extracellular matrix in late senescent human dermal fibroblasts reveals a down-regulated fibronectin-centered network
Journal of Analytical Science and Technology (2022)
-
ALDH7A1 inhibits the intracellular transport pathways during hypoxia and starvation to promote cellular energy homeostasis
Nature Communications (2019)
-
Survive or thrive: tradeoff strategy for cellular senescence
Experimental & Molecular Medicine (2017)
-
Role of AMP-activated protein kinase in cross-talk between apoptosis and autophagy in human colon cancer
Cell Death & Disease (2014)
-
Cannabinoids inhibit energetic metabolism and induce AMPK-dependent autophagy in pancreatic cancer cells
Cell Death & Disease (2013)
