
Abstract
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease caused by the degeneration of motor neurons. Mutations in Cu/Zn superoxide dismutase (SOD1), including G93A, were reportedly linked to familial ALS. SOD1 is a key antioxidant enzyme, and is also one of the major targets for oxidative damage in the brains of patients suffering from Alzheimer's disease (AD). Several lines of evidence suggest that intracellular amyloid beta (Aβ) is associated with the pathogenesis of AD. In this report we demonstrate that intracellular Aβ directly interacts with SOD1, and that this interaction decreases the enzymatic activity of the enzyme. We observed Aβ-SOD1 aggregates in the perinuclear region of H4 cells, and mapped the SOD1 binding region to Aβ amino acids 26-42. Interestingly, intracellular Aβ binds to the SOD1 G93A mutant with greater affinity than to wild-type SOD1. This resulted in considerably less mutant enzymatic activity. Our study implicates a potential role for Aβ in the development of ALS by interacting with the SOD1 G93A mutant.
Similar content being viewed by others


Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease caused by the degeneration of motor neurons. The disorder causes muscle weakness and atrophy throughout the body. In many cases there is a family history of the disease, in which case it is referred to as familial ALS (FALS). Of these, 20% are mapped to the Cu/Zn superoxide dismutase (SOD1) gene on chromosome 21 (Selverstone Valentine et al., 2005).
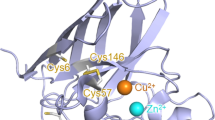
SOD1 is an enzymatic antioxidant found in the cytosol, nucleus, peroxisomes, as well as the mitochondrial intermembrane space of eukaryotic cells and in the periplasmic space of bacteria. The human SOD1 enzyme is a 32-kDa homodimer, having a 153 amino acid subunit including both a copper- and a zinc-binding site. The copper-binding site is the heart of the enzyme active site - the location where the SOD1 protein catalyzes the disproportionation of superoxide to give dioxygen and hydrogen peroxide.
The autosomal dominant nature of SOD1-associated FALS suggests a toxic gain of function for a mutant form of SOD1, and the pioneering work conducted using FALS transgenic mice provided strong support for this hypothesis. Mice over expressing either the human FALS SOD1 G93A mutant or human wild-type SOD1 both showed elevated levels of SOD activity relative to non-transgenic mice, but only the FALS SODI G93A mutant mice developed ALS. To date, however, the means by which the SOD1 mutant exerts its effects remains unclear.
SOD1 is also one of major targets of oxidative damage in the brains of Alzheimer's disease (AD) patients. The amyloid precursor protein (APP), presenilin-1 (PS-1; a component of the secretase complex) and PS-2 are related with early onset AD. Recent reports have demonstrated an intraneuronal accumulation and deposit of amyloid beta (Aβ) in AD-vulnerable brain regions and in brain regions of transgenic mice expressing familial AD (FAD) proteins (Tienari et al., 1997; Gouras et al., 2000; Kienlen-Campard et al., 2002; Wirths et al., 2004). The Aβ peptide is generated by the endoproteolytic processing of a large type 1 transmembrane domain, while the APP is processed to Aβ by β- and γ-secretases (Kogel et al., 2005).
Intracellular Aβ peptides can induce mitochondrial dysfunction and the translocation of cell death-related proteins, such as Smac/DIABLO and Cytochrome C (Canevari et al., 1999; Yin et al., 2002; Park et al., 2004, 2006; Ohyagi et al., 2005). These data support the idea that the accumulation of intracellular Aβ is closely correlated with neuronal cell death. However, the exact mechanism of Aβ toxicity is not completely understood.
Here we show that intracellular Aβ directly interacts with SOD1 and that this interaction impairs the enzymatic activity of SOD1. Importantly, intracellular Aβ binds with the SOD1 G93A mutant with greater affinity than with wild-type SOD1. This resulted in significantly less enzymatic activity of the SOD1 G93A mutant in our experiments. Our results suggest that AD and ALS could potentially be targeted by similar therapeutic strategies.
Results
Intracellular amyloid beta causes H4 cell apoptotic cell death
To study the role of intracellular Aβ, we constructed pAβ and pC100, which contain amino acids 1 to 42 of Aβ and C100, respectively. The N-terminal ends of the fragments were tagged with GFP (Figure 1A and 1B). The H4 human neuroglioma cell line was transfected with pAβ or pC100. We observed apoptosis in cells transfected with both pAβ and pC100 (data not shown). To get some insight into the molecular mechanism of the cell death, poly ADP-ribose polymerase (PARP) was used as a marker for apoptosis. Cells expressing Aβ or C100 showed cleaved PARP, indicating apoptosis (Figure 1C). Aβ also promoted the expression of p53, suggesting a p53-dependent apoptotic mechanism. This result is consistent with that of a previous report (Ohyagi et al., 2005). We also investigated epithelial HEK293 cells. Interestingly, PARP cleavage did not occur in HEK293 cells (data not shown), suggesting that Aβ-mediated apoptosis might be a neuronal cell-specific phenomenon.

Intracellular amyloid beta induces cell death in H4 neuroglioma cells. (A) Schematic representation of human APP and its proteolytic fragments. The functional domains of APP695 include an endoplasmic reticulum (ER) targeting sequence (aa 1-36), a mitochondrial targeting sequence (MTS; aa 36-61) and the Aβ region (aa 597-638). α, β and γ represent the α-, β- and γ-secretase cleavage sites, respectively. C100, a 99-residue C-terminal fragment, is a α-secretase-cleaved APP fragment that can undergo amyloidogenic processing by gamma-secretase to generate the Aβ peptide. (B) Aβ and C100 were tagged with GFP. (C) The expression of Aβ and C100 in H4 cells promoted PARP cleavage and p53 expression. Lane 1 (Mock) shows the result when the empty GFP vector was transfected as a control. (D) Aβ and C100 aggregated in the peri-nuclear region and the cytoplasm.
The subcellular localization of Aβ and C100 was investigated by immunofluorescence microscopy. H4 cells were transfected with pAβ or pC100. Because both pAβ and pC100 contained an N-terminal GFP tag, the localization of Aβ and C100 was detected using anti-GFP antibodies. Aβ and C100 aggregated in the perinuclear region in most cells examined in our experiments (Figure 1D).
SOD1 directly interacts with intracellular amyloid beta
SOD1 mutations were reportedly linked to the pathogenesis of familial amyotrophic lateral sclerosis. Because SOD1 is also one of the major targets for oxidative damage in the brains of AD patients, and due to the fact that intracellular Aβ is associated with the pathogenesis of AD, we investigated the relationship between Aβ and SOD1. We subcloned wild type SOD1 (SOD1 wt) and the SOD1 G93A mutant into a flag-tagged pcDNA vector (Figure 2A). To investigate whether intracellular Aβ interacts with SOD1, we used GFP antibodies to carry out co-immunoprecipitation experiments after co-expressing Aβ and either SOD1 wt or the SOD1 G93A mutant in H4 cells. Both SOD1 wt and the SOD1 G93A mutant were immunoprecipitated, indicating that Aβ interacts with SOD1 (Figure 2B). Importantly, the interaction between Aβ and the SOD1 G93A mutant was approximately five times greater than that between Aβ and wild-type SOD1. As expected, Aβ also bound to endogenous SOD1 (closed arrows; Figure 2B).

SOD1 interacts with intracellular amyloid beta. (A) Schematic representation of wild-type SOD1 and the SOD1 G93A mutant. The metal-binding region of SOD1 consists of one copper- and one zinc-binding site that are in close enough proximity to share an imidazolate ligand His63. The copper ion binding components (His46, His48 and His120) and zinc ion binding components (His71, His80 and Asp83) are involved in the formation of a hydrogen-bonding network in the functionally important regions of SOD1. Mutant G93A corresponds to SOD1 harboring a mutation in which the glycine 93 residue is replaced by an alanine. (B) Interaction between intracellular amyloid beta and wild-type SOD1 or the SOD1 G93A mutant. Wild-type SOD1 is also endogenously expressed in H4 cells (▶, endogenous SOD1; ▷, Flag-tagged wild-type SOD1 or mutant SOD1 G93A. (C) Synuclein and NAC (Non Aβ Component of AD Amyloid), tagged with LexA, do not interact with wild-type SOD1 or the SOD1 G93A mutant. (D) Amyloid beta (green) colocalizes with both SOD1 wild type and the SOD1 G93A mutant (red) when co-expressed in HEK293 cells.
Several neurodegenerative diseases, including Alzheimer's disease and Parkinson's disease, form protein aggregates(Kim et al., 2007; Irvine et al., 2008). To determine whether the SOD1 G93A mutant interacts with proteins that are related to other neurodegenerative disorders, we examined alpha-synuclein and NAC, which are found in the inclusion bodies of patients with Parkinson's disease (Kruger et al., 2000). We determined that neither protein immunoprecipitated with the SOD1 G93A mutant in our experiments (Figure 2C).
To examine the localization of Aβ and SOD1 in cells, we expressed both proteins in HEK293 cells. For co-localization experiments, we used HEK293 cells because HEK293 cells were more resistant to transfection with Aβ or SOD1 G93A than H4 cells. The distribution of the molecules was determined using immunofluorescence microscopy. Aβ and SOD1 colocalized to aggregates in the perinuclear region (Figure 2D). We observed a greater density of aggregates in cells expressing Aβ and the SOD1 G93A mutant compared with cells containing SOD1 wt and Aβ (data not shown). Taken together, these results indicate that Aβ and wild-type SOD1 or the SOD1 G93A mutant interact and colocalize in cells, and that the interaction between Aβ and SOD1 G93A mutant is greater than that between Aβ and wild-type SOD1.
Identification of the SOD1 binding region in amyloid beta
We used specific Aβ domain constructs and in vitro binding assays to determine the region of Aβ that interacts with SOD1. The constructs included pAβ1-16 (amino acids 1-16), pAβ17-42 (amino acids 17-42), pAβ17-25 (amino acids 17 to 25) and pAβ26-100 (amino acids 16-100; Figure 3A). These constructs were modified from our pAβ or pC100 vectors. Because Aβ26-42 was not expressed by mammalian cells, we focused on pAβ26-100 instead. Aβ, Aβ17-42 and Aβ26-100 interacted with both wild-type SOD1 and the SOD1 G93A mutant (Figure 3B and 3C). All these domain constructs interacted with the SOD1 G93A mutant to a greater degree than with wild-type SOD1. These results demonstrate that the Aβ26-42 domain is sufficient for recruiting SOD1.

Aβ amino acids 26-42 interact with SOD1. (A) Schematic representation of the Aβ fragments used. (B) Co-immunoprecipitation of wild-type SOD1 with the Aβ fragments was carried out using H4 cells. (C) Co-immunoprecipitation of the SOD1 G93A mutant and the Aβ fragments was also carried out in H4 cells. ▸, endogenous SOD1; ▹, Flag-tagged wild-type SOD1 or the SOD1 G93A mutant.
Amyloid beta decreases the enzymatic activity of SOD1
The SOD1 protein is an enzyme antioxidant that catalyzes the disproportionation of superoxide to give dioxygen and hydrogen peroxide. We examined if the interaction of SOD1 with Aβ affected its enzymatic activity. To eliminate the effect of endogenous wild-type SOD1, we immuneprecipitated the SOD1-FLAG proteins using a FLAG antibody before evaluating the enzymatic activity of SOD1. The mock vector containing GFP appeared to reduce the expression of SOD1 or the SOD1 G93A mutant. The reduction might be caused by competition for transcription and translation machinery between GFP and SOD1. First of all, we examined whether Aβ reduces the enzymatic activity of the endogenous SOD1 after its immunoprecipitation by an anti-SOD1 antibody (lanes 1 and 2; Figure 4). As expected, the activity of the endogenous SOD1 protein was reduced by the transfected Aβ (lane 2; Figure 4). Similarly, Aβ reduced the enzymatic activity of transfected wild-type SOD1-FLAG proteins (lane 5; Figure 4). Previous reports demonstrated that the SOD1 activity of the SOD1 G93A mutant is at least equivalent to that of wild type SOD1 (lane 6; Figure 4). Our results also indicated that the SOD1 G93A mutant is a functional enzyme (lane 7; Figure 4). However, the SOD1 activity of the SOD1 G93A mutant was greatly reduced by Aβ compared to what we observed for wild-type SOD1 (lane 8; Figure 4). Taken together, our results show that Aβ directly interacts with SOD1 and that this interaction impairs SOD1 enzymatic activity. Our data also indicate that Aβ binds better to the SOD1 G93A mutant than to wild-type SOD1, and that the mutant is affected by the interaction to a greater extent than the wild-type enzyme.

Interaction between SOD1 and intracellular Aβ decreased the enzymatic activity of SOD1. The activity of SOD1 was measured after selecting one type SOD1 following immunoprecipitation. The empty GFP vector alone was transfected as a control. ▸, endogenous SOD1; ▹, Flag-tagged wild-type SOD1 or the SOD1 G93A mutant. All results were performed in triplicate. The error bars indicate standard deviations. *P < 0.05 versus the control (Student's t-test).
Discussion
Our study demonstrates that intracellular Aβ interacts with SOD1. Notably, we observed that Aβ interacts with the SOD1 G93A mutant to a greater degree than to wild-type SOD1. The Aβ-SOD1 interaction impaired SOD1 enzymatic activity, and therefore increased intracellular oxidative stress. Interestingly, we detected apoptotic features in human neuroglioma H4 cells, but not in human epithelial kidney 293 cells. Although the difference between HEK293 cells and H4 cells is still obscure, differences in cell type specificities may offer clues to the apoptotic mechanism caused by the accumulation of Aβ and C100 (Tienari et al., 1997).
Recently, Li et al. (2006) reported that Aβ accelerates the onset of motor impairment and SOD1 aggregation in a murine amyotrophical lateral sclerosis model. These results support our hypothesis that the interaction of Aβ with SOD1 impairs SOD1 enzymatic activity, resulting in apoptotic cell death. Furthermore, these findings suggest that Aβ may directly interact with SOD1 in vivo, including mouse.
Oxidative stress plays an important role in neuronal degeneration in AD. Soluble Aβ, Aβ fibrils, neurofibrillary tangle (NFT), mitochondria abnormalities, and aging all contribute to the increased oxidative stress observed in AD (Chauhan and Chauhan, 2006). SOD1 is a key enzyme needed for ROS catalysis. We found that the direct interaction of Aβ and SOD1, as well as the SOD1 G93A mutant, reduced SOD1 activity. The ROS-mediated oxidative damage of proteins and nucleic acids might be an important event in AD pathology. Because we observed that the SOD1 G93A mutant-Aβ interaction was greater than the wild-type SOD1-Aβ interaction, the activity of the mutant enzyme was consequently affected to a larger degree. Our findings directly correlate with a previous report suggesting that increased Aβ production is associated with SOD1 G93A mutant-linked FALS, and that this may play a role in ALS pathogenesis (Higgins et al., 2003; Li et al., 2006). Our study suggests that AD and ALS might share a common pathogenic mechanism, and that these conditions may potentially be targeted by similar therapeutic strategies.
Methods
Cell culture, transfection and antibodies
Human embryonic kidney (HEK) 293 cells and human neuroglioma H4 cells (American Type Culture Collection) were maintained at 37℃ in a humidified 5% CO2 incubator. The cells were cultured in DMEM supplemented with 8% (v/v) heat-inactivated FBS, 50-units/ml penicillin and 50-µg/ml streptomycin. To introduce the plasmids into HEK293 or H4 cells, the PEI transfection method was used in accordance with the manufacturer's instructions (Sigma). The cells were then incubated for 20 to 30 h at 37℃ in a 5% CO2 incubator. The cells were harvested for various assays. Immunoblots were probed with anti-GFP, anti-SOD1 and anti-LexA antibodies (Santa Cruz Biotechnology); anti-P-JNK1/2 and anti-JNK antibodies (Cell Signaling); anti β-actin and anti-Flag antibodies (Sigma); as well as an anti-PARP antibody (Pharmingen).
Plasmid construction
Total RNA was extracted from lymphocytes and first-strand cDNA was generated with reverse transcriptase. cDNA fragments for Aβ and the C-terminal 100 amino acids (C100) of APP were amplified by PCR with Pfu polymerase (Stratagene) using the first-strand cDNAs as templates. The following primers were used: 5'-GCGCTCGAGCCGGATCCGCCATGGATGCAGAATTC-3' (forward primer for Aβ and C100); 5'-GCGCAAGCTTGTCGACCTACGCTATGACAACAACC-3' (reverse primer for Aβ); and 5'-GCGCAAGCTTGTCGACCTAGTTCTGCATCTGCTC-3' (reverse primer for C100). The amplified fragments were inserted into the pcDNA3.0 GFP vector and the resulting constructs for Aβ and C100 were designated as pAβ and pC100, respectively. The sequence integrity of the constructs was verified by DNA sequencing. pGFP-Aβ 1-16, 17-25, 17-42, 26-100 plasmids were generated by PCR using the pAβ and pC100 constructs.
The cDNA fragments for SOD1 were amplified by PCR with Pfu polymerase (Stratagene) using first-strand human cDNAs as templates. The following primers were used; 5'-GCGCGAATTCATGGCGACGAAGGCCGTGTGC-3' and 5'-GCGCGGTACCTTGGGCGATCCCAATTACACCA-3'. The pcDNA-Flag-SOD1 construct was generated by inserting the amplified fragments into the pcDNA3.0-Flag vector. The QuickChange Site Directed Mutagenesis Kit (Stratagene) was used to substitute the glycine in amino acid position 93 with alanine.
Co-immunoprecipitation assays
For co-immunoprecipitation, transfected HEK293 cells were lysed in lysis buffer (0.2% digitonin, 20 mM HEPES (pH 7.5), 100 mM KCl, 10 mM CaCl2, 50 mM MgCl2 and 1 mM DTT) containing protease inhibitors (2 µg/ml of each aprotinin and leupeptin, with 0.1 mM PMSF) and incubated for 1 h on ice. The protein extracts (1 mg) were precleared with Protein G Sepharose 4 Fast Flow (Amersham Biotechnology) by rocking for 1 h at 4℃, followed by an incubation with anti-GFP (1 µg/ml) or anti-LexA (1 µg/ml) antibodies for 3 h at 4℃. The immunocomplexes were collected by centrifugation after incubating with Protein G Sepharose 4 Fast Flow for 2 h at 4℃. The precipitated Sepharose beads were washed thee times with PBS, and the immunoprecipitated products were eluted from the beads by boiling in sample buffer (60 mM Tris-HCl (pH 6.8), 25% glycerol, 2% SDS, 14.4 mM β-mercaptoethanol and 0.1% bromophenol blue). The eluted proteins were analyzed by 15% SDS-PAGE followed by immunoblot analyses with the anti-SOD1, anti-GFP and anti-LexA antibodies.
The blots were blocked for 1 h at room temperature in 5% non-fat dried milk in TBST (10 mM Tris-HCl (pH 7.5), 150 mM NaCl and 0.1% Tween-20). The membranes were incubated with primary antibodies for 1 h or 3 h, followed by processing with secondary antibodies for 1 h. The antigen-antibody complex was detected using an enhanced chemiluminescence (ECL) kit (Santa Cruz Biotechnology).
Immunofluorescence
Transfected cells were washed with PBS, fixed in 4% formaldehyde for 5 min at room temperature, and then rinsed briefly with PBS. The cells were quenched for 10 min in PBS containing 50 mM NH4Cl and washed three times with PBS. To permeablize the cells, they were incubated for 5 min in PBS containing 0.1% Triton X-100 and were then washed with PBS. The cells were blocked for 1 h in PBS containing 2% BSA. After blocking, the cells were incubated with anti-Flag (1:1000) in blocking solution for 3 h, and then Cy3-coupled anti-mouse IgG (1:500) for 2 h. After washing with blocking solution, the cells were washed with PBS for 5 min and incubated with 2 µg/ml DAPI for 5 min. The subcellular localization of GFP-Amyloid β and SOD1 was analyzed by fluorescence microscopy (Carl Zeiss).
Measurement of SOD activity
Transfected H4 cells were incubated for 10 min at 4℃ in lysis buffer (20 mM HEPES (pH 7.2), 1 mM EGTA, 210 mM mannitol and 70 mM sucrose). The cell pellets were lysed by sonication for 1 min (35%, on-off time; 2 s). The lysates were collected by centrifugation and immmunoprecipitated using anti-Flag or anti-SOD1. The SOD1 activity assay was performed using the Superoxide Dismutase Assay Kit II (Calbiochem) according to the manufacturer's instructions.
Abbreviations
- AD:
-
alzheimer's disease
- Aβ:
-
amyloid beta
- ALS:
-
amyotrophic lateral sclerosis
- H4:
-
human neuroglioma H4
- SOD1:
-
Cu/Zn superoxide dismutase
References
Canevari L, Clark JB, Bates TE . beta-Amyloid fragment 25-35 selectively decreases complex IV activity in isolated mitochondria . FEBS Lett 1999 ; 457 : 131 - 134
Chauhan V, Chauhan A . Oxidative stress in Alzheimer's disease . Pathophysiology 2006 ; 13 : 195 - 208
Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR . Intraneuronal Abeta42 accumulation in human brain . Am J Pathol 2000 ; 156 : 15 - 20
Higgins CM, Jung C, Xu Z . ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes . BMC Neurosci 2003 ; 4 : 16 -
Irvine GB, El-Agnaf OM, Shankar GM, Walsh DM . Protein aggregation in the brain: the molecular basis for Alzheimer's and Parkinson's diseases . Mol Med 2008 ; 14 : 451 - 464
Kienlen-Campard P, Miolet S, Tasiaux B, Octave JN . Intracellular amyloid-beta 1-42, but not extracellular soluble amyloid-beta peptides, induces neuronal apoptosis . J Biol Chem 2002 ; 277 : 15666 - 15670
Kim HJ, Im W, Kim S, Kim SH, Sung JJ, Kim M, Lee KW . Calcium-influx increases SOD1 aggregates via nitric oxide in cultured motor neurons . Exp Mol Med 2007 ; 39 : 574 - 582
Kogel D, Schomburg R, Copanaki E, Prehn JH . Regulation of gene expression by the amyloid precursor protein: inhibition of the JNK/c-Jun pathway . Cell Death Differ 2005 ; 12 : 1 - 9
Kruger R, Muller T, Riess O . Involvement of alpha-synuclein in Parkinson's disease and other neurodegenerative disorders . J Neural Transm 2000 ; 107 : 31 - 40
Li QX, Mok SS, Laughton KM, McLean CA, Volitakis I, Cherny RA, Cheung NS, White AR, Masters CL . Overexpression of Abeta is associated with acceleration of onset of motor impairment and superoxide dismutase 1 aggregation in an amyotrophic lateral sclerosis mouse model . Aging Cell 2006 ; 5 : 153 - 165
Ohyagi Y, Asahara H, Chui DH, Tsuruta Y, Sakae N, Miyoshi K, Yamada T, Kikuchi H, Taniwaki T, Murai H, Ikezoe K, Furuya H, Kawarabayashi T, Shoji M, Checler F, Iwaki T, Makifuchi T, Takeda K, Kira J, Tabira T . Intracellular Abeta42 activates p53 promoter: a pathway to neurodegeneration in Alzheimer's disease . FASEB J 2005 ; 19 : 255 - 257
Park HJ, Seong YM, Choi JY, Kang S, Rhim H . Alzheimer's disease-associated amyloid beta interacts with the human serine protease HtrA2/Omi . Neurosci Lett 2004 ; 357 : 63 - 67
Park HJ, Kim SS, Seong YM, Kim KH, Goo HG, Yoon EJ, Min do S, Kang S, Rhim H . Beta-amyloid precursor protein is a direct cleavage target of HtrA2 serine protease. Implications for the physiological function of HtrA2 in the mitochondria . J Biol Chem 2006 ; 281 : 34277 - 34287
Selverstone Valentine J, Doucette PA, Zittin Potter S . Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis . Annu Rev Biochem 2005 ; 74 : 563 - 593
Tienari PJ, Ida N, Ikonen E, Simons M, Weidemann A, Multhaup G, Masters CL, Dotti CG, Beyreuther K . Intracellular and secreted Alzheimer beta-amyloid species are generated by distinct mechanisms in cultured hippocampal neurons . Proc Natl Acad Sci USA 1997 ; 94 : 4125 - 4130
Wirths O, Multhaup G, Bayer TA . A modified beta-amyloid hypothesis: intraneuronal accumulation of the beta-amyloid peptide--the first step of a fatal cascade . J Neurochem 2004 ; 91 : 513 - 520
Yin KJ, Lee JM, Chen SD, Xu J, Hsu CY . Amyloid-beta induces Smac release via AP-1/Bim activation in cerebral endothelial cells . J Neurosci 2002 ; 22 : 9764 - 9770
Acknowledgements
This work was supported by the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korean Government [R01-2007-000-20032-0]; and Korea Ministry of Health and Welfare (A080588-6).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Yoon, E., Park, HJ., Kim, GY. et al. Intracellular amyloid beta interacts with SOD1 and impairs the enzymatic activity of SOD1: implications for the pathogenesis of amyotrophic lateral sclerosis. Exp Mol Med 41, 611–617 (2009). https://doi.org/10.3858/emm.2009.41.9.067
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2009.41.9.067
Keywords
This article is cited by
-
TDP-43 interacts with amyloid-β, inhibits fibrillization, and worsens pathology in a model of Alzheimer’s disease
Nature Communications (2020)
-
The self-interaction of native TDP-43 C terminus inhibits its degradation and contributes to early proteinopathies
Nature Communications (2012)
-
Two cases of dementias with motor neuron disease evaluated by Pittsburgh compound B-positron emission tomography
Neurological Sciences (2012)
