
Abstract
Osteoblasts can synthesize the insulin-like growth factors (IGFs) and the IGF-binding proteins (IGFBPs), which may either enhance or attenuate IGF-stimulated bone cell proliferation. Since estrogen induced osteoblastic differentiation and proliferation through an estrogen-responsive gene in target cells, we investigated the effects of estrogen on IGFBP-6 expression in the human osteoblastic-like cell line SaOS-2. Expressions of IGFBP-6 protein and mRNA increased 2.8 and 2-fold, respectively, in the presence of 17-β-estradiol (E2) (0.01 to 1 µM) and estrogen receptor (ER) in SaOS-2 cells. On the other hand, E2 induced a 2-fold increase in SaOS-2 cell proliferation. To identify genomic sequences associated with estrogen responsiveness, the 5'-promoter region (-44 to +118) of the IGFBP-6 gene was cloned into a chloramphenicol acetyltransferase (CAT) reporter vector. E2 induced a 3-fold increase in CAT activity in SaOS-2 cells transiently transfected with this construct. Identification of the estrogen-responsive element (ERE) [5'-CCTTCA CCTG-3'] (-9 to +1) in this IGFBP-6 gene promoter region was confirmed using electromobility shift assays and deletion analysis. This functional ERE was important for E2-induced trans-activation of the IGFBP-6 gene. These results demonstrate that E2 exhibits a positive effect on IGFBP-6 gene transcription through estrogen-liganded ER binding to the functional ERE in the IGFBP-6 gene promoter in SaOS-2 cells.
Similar content being viewed by others


Introduction
Insulin-like growth factors (IGFs) play an important role in the regulation of cell metabolism. The biological activity of IGFs is modulated by a family of seven high-affinity IGF binding proteins (IGFBP 1-7) and their receptors (Jones et al., 1995; Oh et al., 1996). Among them, IGFBP-6 has a 20- to 100-fold higher binding affinity for IGF-II than for IGF-I. IGFBP-6 is expressed in many cell types, such as fibroblasts, myoblasts, smooth muscle cells, keratinocytes, and osteoblasts (Yan et al., 2001; Strohbach et al., 2008). Because of its binding preference, IGFBP-6 might regulate IGF-II-induced cell proliferation, differentiation, and survival (Bach et al., 1994; Dake et al., 2002; Kim et al., 2002; Gallicchio et al., 2003; Fu et al., 2007). Generally, IGFBPs play an IGF-dependent role in high affinity IGF binding. Some studies have suggested that IGFBP-6 has IGF-independent effects (Grellier et al., 1998; Seurin et al., 2002; Sueoka et al., 2000). Among the IGFBPs, IGFBP-6 is unique in stimulation of osteosarcoma cell growth and has an anti-apoptotic effect (Schmid et al., 1999). Furthermore, IGFBP-6 inhibits programmed cell death in non-small cell lung cancer cells (Sueoka et al., 2000). Overexpression of IGFBP-6 inhibits both neuroblastoma growth in vivo (Bienvenu et al., 2004) and proliferation of human bronchial epithelial cells (Sueoka et al., 2000). Therefore, IGFBP-6 exerts differential effects on the regulation of cell metabolism in different cell lines.
As an important endocrine-hormone, estrogen is essential for regulation of the growth and differentiation of target cells. Estrogen receptor (ER), a member of the steroid-thyroid hormone receptor superfamily, mediates the action of estrogen by dependently binding ligands to estrogen-responsive elements (EREs) in the target gene promoter, thus directly regulating gene transcription (Evans et al., 1988; Green et al., 1988). ER has been found in female organs and in non-reproductive tissues of the central nervous system (Brown et al., 1988; Simerly et al., 1990), the cardiovascular system (Orimo et al., 1993; Stumpf et al., 1977), and the skeletal system (Eriksen et al., 1988; Komm et al., 1988). It has recently been suggested that estrogen and ER exert differential effects on the biological activity of IGFs. IGFBP-4 was up-regulated by estrogen in the ECC-1 cell line, and IGFBP-6 was down-regulated by progesterone in the PRAB-36 cell line (Gielen et al., 2006). IGFBP-3 and IGFBP-5 were regulated by estrogen in an endometrial cancer cell line (Hondermarck et al., 2003; Gielen et al., 2006). Estrogen (0.01 µM estradiol) reduced the level of IGFBP-3, but induced increase in the level of IGFBP-6 1.5- to 2-fold in MCF-7 cells (Salahifar et al., 2000; Brockdorff et al., 2003). However, the mechanisms of estrogen regulation of IGF signaling are poorly understood. There are few identified EREs in the promoter regions of estrogen-regulated IGF genes.
Structural and functional analyses of the IGFBP-6 gene promoter have been performed (Hodzic et al., 1997; Dailly et al., 2001), and the 5'-promoter region contains retinoic acid response elements, CAAT boxes, CACCC boxes, NF-1, and activated protein-1 (AP-1)/AP-2 sites (Dailly et al., 2001; Song et al., 2001). However, the molecular mechanism of estrogen signaling through ER in IGFBP-6 transcription has not been determined. In order to understand the mechanisms of estrogen in IGF signaling, we previously investigated estrogen regulation of IGFBP-6 gene in vivo (Guo et al., 2003, 2007). Herein, we present evidence that the human osteoblastic-like cell line SaOS-2 expresses IGFBP-6 and that estrogen increases the abundance of both IGFBP-6 mRNA and protein in the presence of ER. We first determined that estradiol exhibits a positive effect on IGFBP-6 gene transcription through a functional ERE in the IGFBP-6 gene promoter region. These results contribute to a better understanding of the transcriptional estrogen regulation of IGFBP-6 gene.
Results
The effect of up-regulation of E2 on IGFBP-6 gene transcription
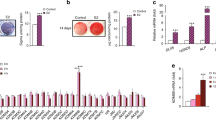
In order to determine whether transcription and translation of the IGFBP-6 gene are regulated by E2, we isolated total mRNA and protein from SaOS-2 cells previously treated with E2 and pSG5HEO (the ERα expression vector). As shown in Figures 2 and 3, E2 induced a significant increase in IGFBP-6 mRNA and protein levels based on RT-PCR and Western blot analytical results. As seen in Figure 2, 1 µM E2 caused a significant 2-fold decrease in the IGFBP-6 mRNA level. As shown in Figure 3, 1 µM E2 resulted in a significant 2.8-fold increase in the IGFBP-6 protein level in SaOS-2 cells, compared with cells untreated with E2. These results indicate that IGFBP-6 gene expression is regulated by E2 at the transcriptional level in vitro. However, at doses of 0.01-1 µM, E2 did not have a dose-dependent effect on IGFBP-6 mRNA expression (Figure 2). Co-treatment with E2 (0.01-1 µM) and ERα caused a significant increase in cell proliferation in SaOS-2 cells compared to treatment with E2 or ERα alone (Figure 1).

Effect of E2 on up-regulation of IGFBP-6 expression in SaOS-2 cells. SaOS-2 cells were cotreated for 24 h with E2 (0.01, 0.1, and 1 µM) in SaOS-2 cells transfected with pSG5HEO. Total RNA and protein from fetal SaOS-2 cells were subjected to semi-quantitative RT-PCR. The band intensity from each sample was quantified using Fluorchem 2.0 software. The level of IGFBP-6 mRNA is presented as a multiple of the β-actin density. RT-PCR results indicated that the level of IGFBP-6 mRNA in SaOS-2 cells treated with 1 µM E2 and pSG5HEO was 2-fold higher than with 1 µM E2 or pSG5HEO alone. *indicates a significant difference between results with and without E2 and/or ER (P < 0.05).

E2 regulation of IGFBP-6 protein level in SaOS-2 cells. SaOS-2 cells were cotreated for 24 h with E2 (0.01, 0.1, and 1 µM) in cells transfected with pSG5HEO. Western blot analysis was used to examine the protein expression of IGFBP-6. The band intensity was quantified using Fluorchem 2.0 software. The level of the IGFBP-6 protein is presented as a multiple of the GAPDH density. Western blot analysis indicated that the level of the IGFBP-6 protein in SaOS-2 cells treated with E2 (0.01, 0.1, 1 µM) and pSG5HEO was 2.8- to 3.5-fold higher than with E2 or pSG5HEO alone. *indicates a significant difference between results with and without E2 and/or ER (P < 0.05).

Estrogen affected SaOS-2 cell proliferation. To determine the effect of estrogen on osteoblastic cell growth in vitro, SaOS-2 cell proliferation was examined using MTT. E2 induced a 2-fold increase in cell proliferation in SaOS-2 cells co-treated with the wild-type human ERα expression vector pSG5HEO. *indicates a significant difference between results with and without E2 or ER (P < 0.05).
Identification of the ERE located in the IGFBP-6 gene promoter
Sequence analysis was carried out to search for EREs in the 1.8 kb region of the human IGFBP-6 gene promoter (GenBank access numbers AF-297519). We did not find EREs in the IGFBP-6 gene promoter using TESS, but the two putative EREs in the IGFBP-6 gene promoter were identified. The putative ERE sequences were 5'-CCTTCACCTG-3' (-9 to +1) and 5'-AACCCTGACC-3' (+105 to +115). To determine whether the identified activated ER-protein can bind to these putative EREs in the IGFBP-6 gene promoter, direct and competitive binding of both the wild-type and the mutant-type ERE oligonucleotides with nuclear extracts was determined using EMSA (Figure 4 and 5). The efficient retarded band appeared after addition of the labeled ERE-1 probe, indicating that the activated ER-protein interacts directly with this natural ERE-1 (Figure 4, Lane 1). In competitive binding studies, this retarded band was subjected to competition with a 50 and 100-fold excess of the unlabeled ERE-1 probe (Figure 4, Lane 4, 5). Addition of the nonspecific labeled probe (mutant-type ERE-1 oligonucleotides) did not result in binding (Figure 4, Lane 6). A similar retarded band did not appear when the labeled wild-type ERE-2 probe was incubated with nuclear extracts (Figure 5, Lane 2). To identify the specific ER in DNA-ER complexes, the ERα antibody was used in EMSA. As shown in Figure 4, the ERα antibody did bind specifically to labeled DNA-ER complexes and formed a supershifted band (Figure 4, Lane 2).

Binding of ER to the identified ERE-1 in the GFBP-6 gene (EMSA). Nuclear extracts from SaOS-2 cells were incubated with a 32P-labeled probe harboring the ERE-1 (Lane 1-5) of the IGFBP-6 gene. The resulting complexes were resolved using non-denaturing PAGE. A labeled mutant-type ERE-1 probe was used as a negative control (Lane 6). For competition EMSA, 50-fold and 100-fold excesses of the unlabeled probe harboring the wild-type ERE-1 (Lane 4, 5) of the IGFBP-6 gene promoter were added during the pre-incubation period. Responsive banding (see arrow in Figure 4) of the wild-type ERE-1 binding complex was observed. Addition of a cold consensus ERE-1 (Lane 4, 5) decreased or eliminated the formation of specific complexes. The retarded band of the mutant-type ERE-1 binding complex was not observed when labeled mutant-type ERE-1 was incubated with nuclear extracts (Lanes 6). The ERα antibody did bind to the putative ERE-1-ER complexes. These specific complexes (Lane 3) were delayed and formed a supershifted band, compared with the ERE-1-ER complexes (Lane 2). The arrow indicates ER binding.

Identification of ERE-2 in the GFBP-6 gene promoter (EMSA). Nuclear extracts from SaOS-2 cells were incubated with a 32P-labeled probe harboring the ERE-2 (Lane 1, 2) of the IGFBP-6 gene. The labeled mutant-type ERE-2 probe was used as a negative control (Lane 3). A 32P-labeled probe harboring ERE-1 (Lane 1-5) was used as a positive control (Lane 5). Responsive banding (see arrow in Figure 5) of the wild-type ERE-1 binding complex was observed (Lane 5). The retarded band of the wild-type and mutant-type ERE-2 binding complex was not observed when a labeled wild-type and mutant-type ERE-2 probe was incubated with nuclear extracts (Lane 2, 3).
Positive effect of E2 on IGFBP-6 gene transcription through the functional ERE located in the IGFBP-6 gene promoter
To examine the function of these two putative EREs, SaOS-2 cells were transiently co-transfected with two constructed pCAT vectors (containing these putative EREs) uisng an ERα expression vector. As shown in Figure 6, 1 µM E2 significantly stimulated CAT activity in -44 IGFBP-6-pCAT [containing ERE-1 (-9 to+1) and ERE-2 (+105 to +115)] transfected cells, but not in +29 IGFBP-6-pCAT [containing only ERE-2 (+105 to +115)] transfected cells. Similar to E2 induction of IGFBP-6 mRNA in SaOS-2 cells, 1 µM E2 induced a 3-fold increase in CAT activity in -44 IGFBP-6-pCAT transfected cells compared to cells transfected with +29 IGFBP-6-pCAT (data not shown). Neither ERE-1 nor E2 alone significantly altered CAT activity in -44 IGFBP-6-CAT transfected cells. These results indicate that the functional ERE-1 (-9 to+1) is located in the IGFBP-6 gene promoter. In the presence of ER, E2 up-regulates CAT activity through ERE-1 (-9 to+1) in the IGFBP-6 gene promoter.

Plasmid constructs and CAT activity in transfected SaOS-2 cells. PCR products from the IGFBP-6 gene promoter were inserted upstream from the luciferase gene in the promoterless pCAT-Basic vector. The putative transcription activation sites are indicated as ERE-1 and ERE-2 in a schematic representation of the constructs (above panel). These constructs were cotransfected with the ERα expression vector into SaOS-2 cells. The transfected cells were then treated with E2 (1 µM) for 24 h. Cellular extracts were prepared and the β-gal and CAT activities were then determined. The CAT activity was normalized to β-gal units and is presented as fold of vehicle control. CAT activities are expressed as the mean ± SEM for three separate determinations in each treatment group. Statistical significance of differences between groups was determined. *indicates a significant difference between the ERE construct and the control group.
Discussion
IGFBP-6, a crucial regulator in the IGF system, is involved in osteogenesis and bone function (Schmid et al., 1999; Milne et al., 2001). Proliferation and differentiation of bone marrow mesenchymal cells are associated with IGFBP-6 and other components of the IGF system (Baddoo et al., 2003). IGFBP-6 is involved in biological effects in an IGF-dependent manner. However, there is growing evidence that IGFBP-6 has IGF-independent effects, including induction of apoptosis and modulation of cell migration (Fu et al., 2007). IGFBP-6 mRNA and proteins are induced by estradiol in ER-positive MCF-7 cells (Martin et al., 1995). Our results confirm that IGFBP-6 is an estradiol-responsive gene. Dailly (Dailly et al., 2001) analyzed the promoter activity of the IGFBP-6 gene 5' -flanking region in the SaOS-2 and U-2OS cell lines. Deletion analysis showed the promoted activity of IGFBP-6 gene requires the -1,731 to +154 bp sequence of the gene promoter, and the important elements include a putative RARE, an AP-2 site, a CACCC box, and an Sp1 site (Dailly et al., 2001; Song et al., 2001). We found that estrogen activity in SaOS-2 cells is primarily associated with the -44 to +118 region of the IGFBP-6 gene promoter that contains the liganded-ER binding site in the -9 to +1 region (Figure 2 and 4).
Estrogen is involved in embryogenesis and development of various tissues (Cummings et al., 2000; Ma et al., 2003; Nautiyal et al., 2004). We previously reported that gene expression of IGF-II is positively regulated by estradiol in MC-3T3-E1 cells. Herein, we found that estradiol strongly stimulates gene transcription of IGFBP-6 in SaOS-2 cells and results in respective 2- and 3.5-fold increases in the gene expressions of IGFBP-6 mRNA and protein. Estradiol differentially regulates production of IGFBP-3 and IGFBP-6 in the MCF-7 cell line (Guo et al., 2007; Martin et al., 1995). E2 (10-8~10-10 M) has similar ability to stimulate the induction of collagenase-3 mRNA in UMR 106-01 cells (Partridge et al., 2000). Transfection with either ERα or ERβ causes U2OS cell sensitivity to E2 (Lima et al., 2004; Kallio et al., 2008). We previously reported that E2 (1 µM) does not positively regulate cell proliferation in MC-3T3-E1 cells (Guo et al., 2007). However, herein, SaOS-2 cells were transfected with the wild-type human ERα expression vector and E2 up-regulated cell proliferation 2 fold in SaOS-2 cells, but in dose independent manner within the range of concentration of 0.01 to 1 µM.
Estrogen regulates some gene expression and the activities of proteins in osteoblasts. However, results concerning some of the reported effects are conflicting. Osteoblasts contain both ERα and ERβ estrogen receptors (Arts et al., 1997; Spelsberg et al., 1999). In addition, the level of ERα increases during osteoblastic cell differentiation (Bodine et al., 1998). Stimulation of estrogen-induced gene expression is mediated by the "classical" mechanism, where by estrogen-liganded ER binds directly to the ERE and interacts with coactivator proteins and components of the RNA polymerase II transcription machinery, resulting in either enhanced or inhibited transcription. The consensus ERE contains a palindrome of PuGGTCA motifs separated by 3 bp (Nardulli et al., 1996; Kuntz et al., 1997). By analyzing the human IGFBP-6 gene promoter sequences, we found two putative EREs (from -9 to +1, and from +105 to +115) in the proximal region of the promoter. In the presence of ERE-1 (-9 to +1) in the IGFBP-6 gene promoter, E2 inducted a significant increase in CAT activity in IGFBP-6-pCAT transfected cells (data not shown). Neither ERE-1 nor E2 alone altered CAT activity in IGFBP-6-CAT transfected cells. The results of EMSA using either 32P-labeled consensus ERE-1 (-9 to +1) or ERE-2 (+105 to +115) oligonucleotides (Figure 4 and 5) demonstrated that ERE-1 (-9 to +1) oligonucleotides formed specifically bound ERE-DNA complexes. Co-incubation with ER resulted in a supershifted ER/ERE-DNA retarded band. These results indicate that the functional ERE (-9 to +1) is located in the IGFBP-6 gene promoter. In the presence of ER, E2 up-regulates CAT activity through this ERE in the IGFBP-6 gene promoter. Therefore, estrogen-liganded ER probably binds to the specific functional ERE (-9 to +1) located in the IGFBP-6 gene promoter and up-regulates IGFBP-6 gene transcription.
We have identified ERE (5'-CCTTCACCTG-3', -9 to +1) in the IGFBP-6 gene promoter, which is important for E2 responsiveness. ER-mediated transactivation via this ERE is involved in induction of the IGFBP-6 gene by E2 in SaOS-2 cells. Acting as an ER genomic binding site, this functional ERE in the IGFBP-6 promoter region is required for estrogen regulation of IGFBP-6 gene transcription. We are currently investigating the integrating role of E2 regulation of several E2-responsive genes via other nuclear transcription factors.
Methods
Cell culture and treatment with E2
SaOS-2 cells (human osteoblastic-like cell line) were routinely cultivated as a monolayer in DMEM (Invitrogen, Merelbeke, Belgium) supplemented with 10% FBS (Hyclone, Erebodegem-Aalst, Belgium), 100 IU/ml of penicillin, and 100 µg/ml of streptomycin (Gemini Bio-Products) in an incubator with 5% CO2 at 37℃. For transient transfection, SaOS-2 cells were plated in 6-well plates and grown to 80% confluence, and then transfected with the 0.2 µg of pSG5HEO (the wild-type human ERα expression vector, Invitrogen) and 10 µl of lipofectamine (Invitrogen). After 16 h, the cells were grown in the presence of E2 (1 µM, 0.1 µM, 0.01 µM) for 24 h before harvesting (Cesario et al., 2007, Guo et al., 2007). E2 was dissolved in 1 µM DMSO. The cells were collected after incubation for 24 h.
Measurement of cell proliferation
After an initial overnight incubation, cells were washed and cell proliferation was determined using the MTT (Sigma-Aldrich) assay. The cells were washed with PBS. MTT was diluted using serum-free medium to obtain a concentration of 0.5 mg/ml, which was added to the culture prior to incubation for 3 h. Formazan extraction was performed using isopropanol, and the quantity of protein was determined using an enzyme linked-immunosorbent assay at 492 nm with four individual samples per group.
RT-PCR
Total RNA was prepared from SaOS-2 cells and used as a template for first strand cDNA synthesis using reverse transcriptase (Invitrogen). Primer sets for semi-quantitative PCR were as follows: IGFBP-6 forward primer, 5'-CTCTCCTCCTGCCCTGTTCTA-3'; IGFBP-6 reverse primer, 5'-CATGGTCAGGGTTTGTGCCC-3' (508 bp); β-actin forward primer, 5'-AGAGCTACGAGCTGCCTGAC-3'; β-actin reverse primer, 5'-AGTACTTGCGCTCAGGAGGA -3' (299 bp). The PCR reaction mixture contained 50 µl of the single-stranded cDNA product, 4 units of Vent polymerase, 1 µM of each primer, 400 µM level of each dNTP, 10 mM Tris-HCI (pH 8.3), 50 mM KCI, and 1.5 mM MgCI2 in a final volume of 100 µl. The mixture was amplified for 30 cycles under the following conditions: denaturing at 94℃ for 45 s; primer annealing at 55℃ for 45 s; and primer extension at 72℃ for 45 s. The PCR products were separated using 2% agarose gel electrophoresis with 100 ng/ml of ethidium bromide. Gels were visualized on a transilluminator and photographed using a gel-automatic formatter (GDS8000, Bio-Rad). Band intensities were analyzed using Fluorchem 2.0 software (Alpha Innotech Corporation). The relative amount of each sample was calculated by normalization to the levels of β-actin mRNA.
Western blot analysis
For immunodetection of the IGFBP-6 protein, SaOS-2 cells were treated with 1 µM E2, 0.1 µM E2, or 0.01 µM E2 for 24 h in 25-cm2 flasks, and then harvested via trypsinization followed by resuspension in 10 mM Tris-HCl (pH 7.4), 100 mM NaCl, 0.5% desoxycholic acid, and 1 mM dithiothreitol containing aprotinin (10 µg/ml) at 4℃ for 30 min. Cells were centrifuged at 100,000 r/min for 30 min. The amount of protein was determined at 595 nm using a protein assay kit (BioRad, Munich, Germany). Protein extracts (10 µg/lane) were run on 12% SDS polyacrylamide gel, and separated proteins were blotted onto a nitrocellulose membrane (Amersham, Freiburg, Germany). Unbound sites were blocked overnight at 4℃ in 10 mM Tris and 0.15 M NaCl (pH 7.4; TBS) containing 5% (wt/vol) skim milk powder. Blots were washed three times for 10 min each time with TBS containing 0.05% Tween-20 (TBST) and incubated for 1 h with the polyclonal anti-human IGFBP-6 antibody (1:1000) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). For verification and quantitation of protein loading, stripped membranes were incubated with the GAPDH antibody (1:400) (Lab Vision Corporation, CA). Blots were again washed three times with TBST as described above, and immunoreactive bands were visualized using ECL plus Western blotting detection reagents (Amersham Pharmacia Biotech Inc, Piscataway, NJ). The antibody-bound protein intensities were analyzed using a Chemi-imager-5500 V 2.03 electrophoresis gel image system. The housekeeping gene product GAPDH was used as an internal control for protein integrity. The target proteins are quantified as relative values against GAPDH as an internal reference.
Electromobility shift assays (EMSA)
Nuclear extracts from SaOS-2 cells were prepared using the method of Guo et al. (2007). The concentrations of nuclear extracts were determined according to the Bradford method (Bradford et al., 1976). The putative consensus motifs of EREs are mnCnsTGACC and mrCnnTGACC (m=A, C; n=T, C, G, A) as predicted using Transcription Element Search Software (TESS), while the sequences in a human IGFBP-6 promoter DNA fragment are 5'-CCTTCACCTG-3' (-9 to +1) and 5'-AACCCTGACC-3' (+105 to +115). The initiation site for transcription is marked by a star and numbered as +1. The putative ERE probes (oligonucleotides) in the human IGFBP-6 promoter were synthesized, including the wild-type ERE-1 probe (DNA sequence is 5'-CCACCCCCCTTCACCTGGCTCTTA A-3' and 5'-TTAAGAGCCAGGTGAAGGGG-3') (-15 to +9), the wild-type ERE-2 probe (DNA sequence is 5'-GACGGGGCACAAACCCTGACCATGA-3' and 5'-TCATGGTCAGGGTTTGTGCCCCGTC-3') (+94 to +119), and the mutant-type ERE-1 probe (DNA sequence is 5'-CCACCCCCCTCCGAACTGCTCTTAA-3' and 5'-TTAAGAGCAGTTCGGAGGGGGGTGG-3'). This mutant-type ERE-1 probe was used as a negative control. It does not contain any known binding sequences. The ERE oligonucleotides were chemically synthesized, annealed, and labeled at the 5'-ends using polynucleotide kinase and [γ-32P] d-ATP (3,000 Ci/mmol). The labeled wild-type EREs and the mutant ERE probe were incubated with 5 µg of nuclear extract and 1 µg of poly (dI-dC) in a buffer containing 10 mM HEPES (pH 7.5), 50 mM KCl, 5 mM MgCl2, 0.5 mM EDTA, 1 mM dithiothreitol, and 12.5% glycerol on ice for 90 min. Reaction mixtures were loaded onto 8% polyacrylamide gel (acrylamide/bisacrylamide ratio, 29: 1) and run at 110 V in 0.375 × TBE (0.09 M Tris, 0.09 M borate, 2 mM EDTA, pH 8.3). For competition analysis, 50-fold and 100-fold molar excesses of the cold ERE probe were added to the reaction mixture before addition of the labeled probe. To identify the specific ER in DNA-ER complexes, the ERα antibody (1:1000) (rabbit polyclonal IgG against the C terminus of the ERα, Santa Cruz Biotechnology, CA) was added to the reaction mixture after labeled wild-type EREs were incubated with nuclear extracts for 30 min at 4℃. The protein-nucleic acid complexes were identified using autoradiography for 48 h at -70℃.
Plasmid constructs
The chloramphenicol acetyltransferase (CAT) reporter constracts, -44IGFBP-6-pCAT and +29IGFBP-6-pCAT containing the human IGFBP-6 gene promoter sequences from -44 to +118 and from +29 to +118, respectively, were prepared using PCR-amplified DNA fragments and CAT reporter plasmids derived from pCAT-Basic (Promega).
These two DNA fragments were amplified by PCR with the forward primers 5'-AGTTTAGGGAATGCCCGT-3' and 5'-CGGCTACTTAAGACAGAG-3', and the common reverse primer 5'-CATGGTCAGGGTTTGTGCCC-3' using human genomic DNA as a template. The -44IGFBP-6-pCAT construct contains putative ERE-1 (-9 to +1) and ERE-2 (+105 to +115) located in the promoter region of the IGFBP-6 gene. In contrast, the +29 IGFBP-6-CAT construct contains only putative ERE-2. All constructs were confirmed using nucleotide sequence analysis (Figure 6).
Cell culture and transient cotransfection assays
The SaOS-2 cells were grown in DMEM supplemented with 10% FBS, 100 units/ml of penicillin, and 100 µg/ml of streptomycin. Cells were maintained in a 5% CO2, 95% air humidified atmosphere at 37℃. For transient transfection, SaOS-2 cells were plated in 6-well plates and grown to 80% confluence, and then transfected with 5 µg of the reporter construct (-44 IGFBP-6-pCAT or +29 IGFBP-6-pCAT), 0.2 µg of pSG5HEO (the wild-type human ERα expression vector, Invitrogen) and 10 µl of Lipofectamine (Invitrogen, 11668-027). An amount of 0.2 µg of the pSV-β-Galactosidase vector (Promega) was added as an internal control. After 16 h, the cells were grown in the presence of E2 (1 µM) for 24 h before harvesting. Cell extracts were prepared by three rapid freeze /thaw cycles and the CAT activity was assayed using a CAT-ELISA kit (Roche, Germany). The transfection efficiency was normalized by measuring the β-galactosidase (β-gal) activity in the extracts. The β-gal activity was measured by adding 50 µl of cell lysate to 500 µl of buffer A [100 nM Na2HPO4 (pH 7.2), 10 mM KCl, 1 mM MgCl2, 10 mM β-mercaptoethanol, and 15 mg/ml of β-D-galactoside sodium (Calbiochem, Germany)] and read at O.D.574. CAT activity is presented as fold of vehicle control.
Data analysis
Data are presented as the mean ± SEM. A one-way analysis of variance (ANOVA) followed by the post hoc Tukey-Kramer test was used to determine differences among multiple groups. Student's t-test was used to determine differences between two groups. P<0.05 was defined as the level of statistical significance.
Abbreviations
- E2:
-
17-β-estradiol
- EMSA:
-
electromobility shift assays
- ER:
-
estrogen receptor
- ERE:
-
estrogen-responsive element
- IGFBPs:
-
insulin-like growth factor binding proteins
- IGFs:
-
insulin-like growth factors
References
Arts J, Kuiper GG, Janssen JM, Gustafsson JA, Löwik CW, Pols HA, van Leeuwen JP . Differential expression of estrogen receptors alpha and beta mRNA during differentiation of human osteoblast SV-HFO cells . Endocrinology 1997 ; 138 : 5067 - 5070
Bach LA, Hsieh S, Brown AL, Rechler MM . Recombinant human insulin-like growth factor (IGF)-binding protein-6 inhibits IGF-II-induced differentiation of L6A1 myoblasts . Endocrinology 1994 ; 135 : 2168 - 2176
Baddoo M, Hill K, Wilkinson R, Gaupp D, Hughes C, Kopen GC, Phinney DG . Characterization of mesenchymal stem cells isolated from murine bone marrow by negative selection . J Cell Biochem 2003 ; 89 : 1235 - 1249
Bienvenu G, Seurin D, Grellier P, Froment P, Baudrimont M, Monget P, Le Bouc Y, Babajko S . Insulin-like growth factor binding protein-6 transgenic mice: postnatal growth, brain development, and reproduction abnormalities . Endocrinology 2004 ; 145 : 2412 - 2420
Bodine PV, Henderson RA, Green J, Aronow M, Owen T, Stein GS, Lian JB, Komm BS . Estrogen receptor-alpha is developmentally regulated during osteoblast differentiation and contributes to selective responsiveness of gene expression . Endocrinology 1998 ; 139 : 2048 - 2057
Bradford MM . A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding . Anal Biochem 1976 ; 72 : 248 - 254
Brockdorff BL, Heiberg I, Lykkesfeldt AE . Resistance to different antiestrogens is caused by different multi-factorial changes and is associated with reduced expression of IGF receptor Ialpha . Endocr Relat Cancer 2003 ; 10 : 579 - 590
Brown TJ, Hochberg RB, Zielinski JE, MacLusky NJ . Regional sex differences in cell nuclear estrogen-binding capacity in the rat hypothalamus and preoptic area . Endocrinology 1988 ; 123 : 1761 - 1770
Cesario RM, Stone J, Yen WC, Bissonnette RP, Lamph WW . Differentiation and growth inhibition mediated via the RXR:PPARgamma heterodimer in colon cancer . Cancer Lett 2006 ; 240 : 225 - 233
Cummings AM, Laws SC . Assessment of estrogenicity by using the delayed implanting rat model and examples . Reprod Toxicol 2000 ; 14 : 111 - 117
Dailly YP, Zhou Y, Linkhart TA, Baylink DJ, Strong DD . Structure and characterization of the human insulin-like growth factor binding protein (IGFBP)-6 promoter: identification of a functional retinoid response element . Biochim Biophys Acta 2001 ; 1518 : 145 - 151
Dake BL, Boes M, Bach LA, Bar RS . Effect of an insulin-like growth factor binding protein fusion protein on thymidine incorporation in neuroblastoma and rhabdomyosarcoma cell lines . Endocrinology 2004 ; 145 : 3369 - 3374
Eriksen EF, Colvard DS, Berg NJ, Graham ML, Mann KG, Spelsberg TC, Riggs BL . Evidence of estrogen receptors in normal human osteoblast-like cells . Science 1988 ; 241 : 84 - 86
Evans RM . The steroid and thyroid hormone receptor superfamily . Science 1988 ; 240 : 889 - 895
Fu P, Thompson JA, Bach LA . Promotion of cancer cell migration: an insulin-like growth factor (IGF)-independent action of IGF-binding protein-6 . J Biol Chem 2007 ; 282 : 22298 - 22306
Gallicchio MA, Kaun C, Wojta J, Binder B, Bach LA . Urokinase type plasminogen activator receptor is involved in insulin-like growth factor-induced migration of rhabdomyosarcoma cells in vitro . J Cell Physiol 2003 ; 197 : 131 - 138
Gielen SC, Hanekamp EE, Hanifi-Moghaddam P, Sijbers AM, van Gool AJ, Burger CW, Blok LJ, Huikeshoven FJ . Growth regulation and transcriptional activities of estrogen and progesterone in human endometrial cancer cells . Int J Gynecol Cancer 2006 ; 16 : 110 - 120
Green S, Chambon P . Nuclear receptors enhance our understanding of transcription regulation . Trends Genet 1988 ; 4 : 309 - 314
Grellier P, De Galle B, Babajko S . Expression of insulin-like growth factor-binding protein 6 complementary DNA alters neuroblastoma cell growth . Cancer Res 1998 ; 58 : 1670 - 1676
Guo L, Zhao YY, Dong LY, Ding Q, Sun KL, Ji SJ, Wang LZ . Antiestrogenic effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on the gene expression of insulin-like growth factors family in osteoblast cells . Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2003 ; 20 : 490 - 494
Guo L, Zhao YY, Zhao YY, Sun ZJ, Liu H, Zhang SL . Toxic effects of TCDD on osteogenesis through altering IGFBP-6 gene expression in osteoblasts . Biol Pharm Bull 2007 ; 30 : 2018 - 2026
Hodzic D, Delacroix L, Willemsen P, Bensbaho K, Collette J, Broux R, Lefèbvre R, Legros JJ, Grooteclaes M, Winkler R . Characterization of the IGF system and analysis of the possible molecular mechanisms leading to IGF-II overexpression in a mesothelioma . Horm Metab Res 1997 ; 29 : 549 - 555
Hondermarck H . Breast cancer: when proteomics challenges biological complexity . Mol Cell Proteomics 2003 ; 2 : 281 - 291
Jones JI, Clemmons DR . Insulin-like growth factors and their binding proteins: biological actions . Endocr Rev 1995 ; 16 : 3 - 34
Kallio A, Guo T, Lamminen E, Seppänen J, Kangas L, Väänänen HK, Härkänen P . Estrogen and the selective estrogen receptor modulator (SERM) protection against cell death in estrogen receptor alpha and beta expressing U2OS cells . Mol Cell Endocrinol 2008 ; 289 : 38 - 48
Kim EJ, Kang YH, Schaffer BS, Bach LA, MacDonald RG, Park JH . Inhibition of Caco-2 cell proliferation by all-trans retinoic acid: role of insulin-like growth factor binding protein-6 . J Cell Physiol 2002 ; 190 : 92 - 100
Komm BS, Terpening CM, Benz DJ, Graeme KA, Gallegos A, Korc M, Greene GL, O'Malley BW, Haussler MR . Estrogen binding, receptor mRNA, and biologic response in osteoblast-like osteosarcoma cells . Science 1988 ; 241 : 81 - 84
Kuntz MA, Shapiro DJ . Dimerizing the estrogen receptor DNA binding domain enhances binding to estrogen response elements . J Biol Chem 1997 ; 272 : 27949 - 27956
Lima F, Vico L, Lafage-Proust MH, van der Saag P, Alexandre C, Thomas T . Interactions between estrogen and mechanical strain effects on U2OS human osteosarcoma cells are not influenced by estrogen receptor type . Bone 2004 ; 35 : 1127 - 1135
Ma WG, Song H, Das SK, Paria BC, Dey SK . Estrogen is a critical determinant that specifies the duration of the window of uterine receptivity for implantation . Proc Natl Acad Sci U S A 2003 ; 100 : 2963 - 2968
Martin JL, Coverley JA, Pattison ST, Baxter RC . Insulin-like growth factor-binding protein-3 production by MCF-7 breast cancer cells: stimulation by retinoic acid and cyclic adenosine monophosphate and differential effects of estradiol . Endocrinology 1995 ; 136 : 1219 - 1226
Milne M, Quail JM, Rosen CJ, Baran DT . Insulin-like growth factor binding proteins in femoral and vertebral bone marrow stromal cells: expression and regulation by thyroid hormone and dexamethasone.Department of Orthopedics and Physical Rehabilitation . J Cell Biochem 2001 ; 81 : 229 - 240
Nardulli AM, Romine LE, Carpo C, Greene GL, Rainish B . Estrogen receptor affinity and location of consensus and imperfect estrogen response elements influence transcription activation of simplified promoters . Mol Endocrinol 1996 ; 10 : 694 - 704
Nautiyal J, Kumar PG, Laloraya M . 17Beta-estradiol induces nuclear translocation of CrkL at the window of embryo implantation . Biochem Biophys Res Commun 2004 ; 318 : 103 - 112
Oh Y, Nagalla SR, Yamanaka Y, Kim HS, Wilson E, Rosenfeld RG . Synthesis and characterization of insulin-like growth factor-binding protein (IGFBP)-7. Recombinant human mac25 protein specifically binds IGF-I and -II . J Biol Chem 1996 ; 271 : 30322 - 30325
Orimo A, Inoue S, Ikegami A, Hosoi T, Akishita M, Ouchi Y, Muramatsu M, Orimo H . Vascular smooth muscle cells as target for estrogen . Biochem Biophys Res Commun 1993 ; 195 : 730 - 736
Partridge NC, Fiacco GJ, Walling HW, Barmina OY, Jeffrey JJ, Ruh MF . Effects of dioxin and estrogen on collagenase-3 in UMR 106-01 osteosarcoma cells . Arch Biochem Biophys 2000 ; 382 : 182 - 188
Salahifar H, Baxter RC, Martin JL . Differential regulation of insulin-like growth factor-binding protein-3 protease activity in MCF-7 breast cancer cells by estrogen and transforming growth factor-beta1 . Endocrinology 2000 ; 141 : 3104 - 3110
Schmid C, Keller C, Gosteli-Peter M, Zapf J . Mitogenic and antiapoptotic effects of insulin-like growth factor binding protein-6 in the human osteoblastic osteosarcoma cell line Saos-2/B-10 . Biochem Biophys Res Commun 1999 ; 263 : 786 - 789
Seurin D, Lassarre C, Bienvenu G, Babajko S . Insulin-like growth factor binding protein-6 inhibits neuroblastoma cell proliferation and tumour development . Eur J Cancer 2002 ; 38 : 2058 - 2065
Simerly RB, Chang C, Muramatsu M, Swanson LW . Distribution of androgen and estrogen receptor mRNA-containing cells in the rat brain: an in situ hybridization study . J Comp Neurol 1990 ; 294 : 76 - 95
Song BL, Qi W, Yang XY, Chang CC, Zhu JQ, Chang TY, Li BL . Organization of human ACAT-2 gene and its cell-type-specific promoter activity . Biochem Biophys Res Commun 2001 ; 282 : 580 - 588
Spelsberg TC, Subramaniam M, Riggs BL, Khosla S . The actions and interactions of sex steroids and growth factors/cytokines on the skeleton . Mol Endocrinol 1999 ; 13 : 819 - 828
Strohbach C, Kleinman S, Linkhart T, Amaar Y, Chen ST, Mohan S, Strong D . Potential involvement of the interaction between insulin-like growth factor binding protein (IGFBP)-6 and LIM mineralization protein (LMP)-1 in regulating osteoblast differentiation . J Cell Biochem 2008 ; 104 : 1890 - 1905
Stumpf WE, Sar M, Aumüller G . The heart: a target organ for estradiol . Science 1977 ; 196 : 319 - 321
Sueoka N, Lee HY, Walsh GL, Fang B, Ji L, Roth JA, LaPushin R, Hong WK, Cohen P, Kurie JM . Insulin-like growth factor binding protein-6 inhibits the growth of human bronchial epithelial cells and increases in abundance with all-trans-retinoic acid treatment . Am J Respir Cell Mol Biol 2000 ; 23 : 297 - 303
Sueoka N, Lee HY, Wiehle S, Cristiano RJ, Fang B, Ji L, Roth JA, Hong WK, Cohen P, Kurie JM . Insulin-like growth factor binding protein-6 activates programmed cell death in non-small cell lung cancer cells . Oncogene 2000 ; 19 : 4432 - 4436
Yan T, Wergedal J, Zhou Y, Mohan S, Baylink DJ, Strong DD . Inhibition of human osteoblast marker gene expression by retinoids is mediated in part by insulin-like growth factor binding protein-6 . Growth Horm IGF Res 2001 ; 11 : 368 - 377
Acknowledgements
We thank Dr. S. Shimasaki for providing the IGFBP-6 gene probe for gene transcription analysis. This study was financially supported by the National Natural Science Foundation of China (No. 30872724, 30500414) and the Scientific Research Project of the Department of Education of Liaoning Province (No. 20061010)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zhao, Yy., Guo, L., Zhao, Xj. et al. Transcriptional activation of insulin-like growth factor binding protein 6 by 17β-estradiol in SaOS-2 cells. Exp Mol Med 41, 478–486 (2009). https://doi.org/10.3858/emm.2009.41.7.053
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2009.41.7.053
Keywords
This article is cited by
-
Regulation of insulin-like growth factors and their binding proteins by thyroid stimulating hormone in human osteoblast-like (SaOS2) cells
Molecular and Cellular Biochemistry (2012)
