
Abstract
Serum amyloid A (SAA) has been regarded as an important mediator of inflammatory responses. The effect of several formyl peptide receptor-like 1 (FPRL1) ligands on the production of IL-8 by SAA was investigated in human neutrophils. Among the ligands tested, LL-37 was found to specifically inhibit SAA-induced IL-8 production in transcriptional and post-transcriptional levels. Since SAA stimulated IL-8 production via ERK and p38 MAPK in human neutrophils, we tested the effect of LL-37 on SAA induction for these two MAPKs. LL-37 caused a dramatic inhibition of ERK and p38 MAPK activity, which is induced by SAA. LL-37 was also found to inhibit SAA-stimulated neutrophil chemotactic migration. Further, the LL-37-induced inhibitory effect was mediated by FPRL1. Our findings indicate that LL-37 is expected to be useful in the inhibition of SAA signaling and for the development of drugs against SAA-related inflammatory diseases.
Similar content being viewed by others


Introduction
Serum amyloid A (SAA) is recognized as a major acute-phase protein released into the blood circulation in response to infection or injury (Jensen and Whitehead, 1998; Uhlar and Whitehead, 1999; Urieli-Shoval et al., 2000). According to previous reports, the concentration of SAA has been reported to be 1,000 times the normal level during acute-phase reactions (Jensen and Whitehead, 1998; Uhlar and Whitehead, 1999; Urieli-Shoval et al., 2000). Also, a previous report demonstrated that SAA possesses a number of immunomodulatory roles, including a potent chemoattractant for human leukocytes, including monocytes and neutrophils (Badolato et al., 1994). It has been found that SAA selectively stimulates formyl peptide receptor-like 1 (FPRL1), which results in Ca2+ mobilization and cell migration (Su et al., 1999a). Furthermore, Su et al. (1999a) demonstrated that radiolabeled SAA specifically binds to human phagocytes and 293 FPRL1-transfected cells, thus demonstrating that FPRL1 is a specific SAA receptor. Recently, He et al. (2003) demonstrated that SAA stimulates IL-8 production in human neutrophils by activating FPRL1. Although the production of pro-inflammatory cytokines such as IL-8 is required for the immune responses, excess production of these cytokines elicits some adverse effects, which result in tissue damage and inflammatory disease. Since SAA has been determined to be a crucial mediator for inflammatory responses, researchers have placed more importance on identifying molecules that inhibit SAA-induced cellular responses. The putative molecules that can block SAA's actions are considered very useful for the development of anti-inflammatory therapeutic agents.
In addition to SAA, several molecules including LL-37 (Yang et al., 2000), a mitochondrial peptide fragment MYFINILTL (Chiang et al., 2000), an important lipid mediator (lipoxin A4) (Maddox et al., 1997), some peptides (T21/DP107, F peptide, and V3 peptide) derived from HIV-1 envelope proteins (Deng et al., 1999; Su et al., 1999b), and Trp-Lys-Tyr-Met-Val-D-Met-CONH2 (WKYMVm) (Seo et al., 1997; Le et al., 1999), have been reported as agonists for FPRL1. Previously, it has been demonstrated that FPRL1 can be differentially stimulated to express ligand-selective properties (Chiang et al., 2000; Bae et al., 2003; Lee et al., 2005, 2006). Furthermore, Chiang et al. (2000) reported that the stimulation of FPRL1 by its peptide ligands resulted in the activation of leukocytes; however, the stimulation of the receptor by lipoxin A4 leads to anti-inflammatory functioning. We also demonstrated that FPRL1 could be differentially activated by distinct peptide ligands via superoxide anion generation, matrix-metalloproteinase-9 production, and lipid mediator production (Bae et al., 2003; Lee et al., 2005, 2006). Although FPRL1 activation by each ligand has been studied for several years, the effect of FPRL1 ligands on a strong FPRL1 agonist (SAA)-induced cellular signal has not been examined. In this study, we investigated the effects of several FPRL1 agonists on the SAA-induced IL-8 production in human neutrophils. Further, we investigated the involvement of FPRL1 in LL-37-induced anti-inflammatory responses as well as the signaling pathways involved in this process.
Results
LL-37 inhibits SAA-stimulated IL-8 production in human neutrophils
To investigate the effect of several FPR family ligands on IL-8 production in human neutrophils, we stimulated the cells with each peptide for 24 h. As shown in Figure 1A, SAA was the only peptide tested that induced IL-8 production at 24 h after stimulation. The other peptides had no measurable effect on IL-8 production in human neutrophils (Figure 1A). In a separate experiment, we observed that all the tested FPR family ligands stimulated the chemotactic migration of human neutrophils, indicating that all the FPR family ligands used are active (data not shown). Next, we checked whether other FPR family agonists affected SAA-induced IL-8 production. The preincubation of human neutrophils with an effective concentration (10 µM) of LL-37 prior to the addition of SAA, resulted in the inhibition of the SAA-induced IL-8 production. The combination of the preincubation of human neutrophils and the effective concentrations of the other tested FPR family agonists (aside from LL-37); namely, fMLF (1 µM), MMK-1 (1 µM), WKYMVm (1 µM), and F2L (20 µM) did not affect SAA-induced IL-8 production in cells (Figure 1B). We also found that higher concentrations (10 µM or 20 µM) of other FPRL1 agonists (fMLF, MMK-1, WKYMVm, and F2L), did not inhibit SAA-induced IL-8 production (data not shown).

Effect of several FPRL1 agonists on IL-8 production in human neutrophils. Freshly isolated human neutrophils stimulated by fMLF (1 µM), MMK-1 (1 µM), WKYMVm (1 µM), LL-37 (10 µM), F2L (20 µM), SAA (2 µM) or LPS (100 ng/ml) for 24 h (A). Human neutrophils stimulated by fMLF (1 µM), MMK-1 (1 µM), WKYMVm (1 µM), LL-37 (10 µM), F2L (20 µM), and subsequently stimulated with SAA (2 µM) for 24 h (B). The IL-8 levels secreted were determined by ELISA. The data represent the mean ± SE of three independent experiments performed in duplicate (A, B).
Increasing the concentration of LL-37 prior to the addition of SAA resulted in the inhibition of the SAA-induced IL-8 production in a concentration-dependent manner. Namely, 5-10 µM of LL-37 almost completely inhibited the IL-8 production induced by SAA (Figure 2A). However, the same concentration of LL-37 (10 µM) did not inhibit LPS-induced IL-8 production in human neutrophils (Figure 2B).

LL-37 specifically inhibits SAA-induced IL-8 production in human neutrophils. Human neutrophils were stimulated at various concentrations of LL-37 for 10 min prior to addition of SAA (2 µM) for 24 h (A). Human neutrophils were stimulated with LPS (100 ng/ml) for 24 h in the absence or presence of LL-37 (10 µM) (B). The data represent the mean ± SE of three independent experiments performed in duplicate (A, B). Human neutrophils, stimulated with LL-37 (10 µM), SAA (2 µM), LL-37 (10 µM) + SAA (2 µM), LPS (100 ng/ml), or LL-37 (10 µM) + LPS (100 ng/ml) for 6 h, were harvested for RNA preparation. A RT-PCR was performed using specific primers for human IL-8 and GAPDH. Next, the PCR products were electrophoresed in 2% agarose gel and stained with ethidium bromide. The data obtained from one representative experiment of experiments performed in quadruplicate is shown (C).
LL-37-inhibited IL-8 production is mediated in a transcriptional level
To investigate the mechanism involved in LL-37-inhibited IL-8 production by SAA, we examined the effects of SAA and LL-37+SAA on the accumulation of IL-8 mRNA transcripts by RT-PCR. As shown in Figure 2C, the stimulation of human neutrophils with SAA (2 µM) resulted in IL-8 mRNA transcript accumulation at 6 h after stimulation. However, 10 µM of LL-37 alone did not affect IL-8 mRNA transcript accumulation. The preincubation of LL-37 prior to SAA stimulation, resulted in the complete inhibition of IL-8 mRNA accumulation by SAA (Figure 2C). As a control, we determined that LPS also stimulated IL-8 mRNA accumulation in human neutrophils, which was not inhibited by LL-37 (Figure 2C).
LL-37 inhibits SAA-induced ERK and p38 MAPK activity
MAPK has been reported to mediate extracellular signals into the nucleus, in several cell types (Johnson and Lapadat, 2002). Moreover, several reports also demonstrated that MAPK activity is required for the induction of several proinflammatory cytokines, such as IL-8 (Coxon et al., 2003; Jo et al., 2004). To determine the role of individual MAPK on SAA-induced IL-8 production, we preincubated human neutrophils with PD98059 (a selective MEK inhibitor) or SB203580 (a selective p38 MAPK inhibitor) prior to SAA treatment. As shown in Figure 3A, SAA-induced IL-8 production was dramatically inhibited by both PD98059 and SB203580. Thus, we demonstrated that both MEK-dependent MAPK and p38 MAPK are essential for SAA-induced IL-8 production in human neutrophils.

LL-37 blocks SAA-induced ERK and p38 MAPK activity in human neutrophils. Human neutrophils were preincubated with vehicle (DMSO), PD98059 (50 µM) for 60 min, or SB203580 (20 µM) for 15 min prior to treatment with SAA (2 µM) for 24 h (A). The amount of IL-8 secreted was measured by ELISA. The data represent the mean ± SE of three independent experiments performed in duplicate. Human neutrophils were stimulated with LL-37 (10 µM), SAA (2 µM), or LL-37 (10 µM) + SAA (2 µM) for 5 min. Samples (30 µg of protein) were subjected to 10% SDS-PAGE, and the phosphorylated ERK or phosphorylated p38 MAPK levels were determined by immunoblot analysis using anti-phospho-ERK antibody or anti-phospho-p38 MAPK antibody (B). The results shown are representative of at least three independent experiments (B).
We also tested the effect of SAA on MAPK activity by Western blot analysis. We found that after stimulating the human neutrophils with 2 µM of SAA for 5 min, the phosphorylation level of ERK dramatically increased (Figure 3B). Another important MAPK, p38 MAPK, was also activated by SAA (Figure 3B). However, LL-37 alone did not stimulate ERK or p38 MAPK in human neutrophils (Figure 3B). We also investigated the effect of LL-37 on the SAA-induced MAPK activity. The preincubation of human neutrophils with LL-37 prior to the addition of SAA elicited an almost complete inhibition of SAA-induced ERK phosphorylation (Figure 3B). We also found that LL-37 inhibited SAA-induced p38 MAPK phosphorylation in human neutrophils (Figure 3B).
LL-37-induced inhibitory effect is mediated by FPRL1
The finding which demonstrated that SAA (an FPRL1 agonist) elicited IL-8 induction in human neutrophils, and that LL-37 inhibited this IL-8 production by SAA, led us to examine whether SAA stimulates IL-8 production via FPRL1 in human neutrophils. We investigated the effects of SAA on IL-8 expression in FPRL1-expressing RBL-2H3 cells. As shown in Figure 4A, the stimulation of FPRL1-expressing RBL-2H3 cells with 2 µM SAA elicited IL-8 mRNA accumulation. Moreover, LL-37 alone did not stimulate IL-8 mRNA accumulation in FPRL1-expressing RBL-2H3 cells. Conversely, the preincubation of human neutrophils with LL-37, prior to the addition of SAA, completely blocked SAA-induced IL-8 mRNA accumulation (Figure 4A). In the vector-expressing RBL-2H3 cells, none of the agonists tested were found to stimulate IL-8 mRNA induction (Figure 4B).

Role of FPRL1 on the inhibitory effect of LL-37 against SAA-induced IL-8 production. FPRL1- (A) or vector- (B) expressing RBL-2H3 cells were stimulated with LL-37 (10 µM), SAA (2 µM), or LL-37 (10 µM) + SAA (2 µM) for 6 h and then harvested for RNA preparation. A RT-PCR was performed using specific primers for rat IL-8 and actin. The PCR products were electrophoresed in 2% agarose gel and stained with ethidium bromide. The data reported from one experiment is representative of the four (A, B).
LL-37 inhibits SAA-stimulated chemotactic migration in human neutrophils
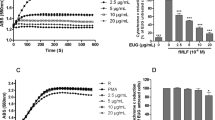
We examined the influence of LL-37 and SAA on the migration of human neutrophils using various concentrations of LL-37 and SAA for the chemotaxis assay. SAA was found to have a concentration dependant influence on neutrophil migration. Further, we found that the maximum human neutrophil migration rate was observed with 1-2 µM of SAA (Figure 5A). However, LL-37 elicited a weak influence on neutrophil chemotaxis. In comparison, LL-37-induced neutrophil chemotaxis activity was less than 10% of the SAA-induced neutrophil chemotaxis activity (Figure 5A). After determining the effect of LL-37 and SAA on the chemotactic migration in human neutrophils, we examined whether LL-37 had an inhibitory effect on SAA-induced neutrophil chemotaxis. The addition of LL-37 caused an almost complete inhibition of SAA-stimulated neutrophil chemotaxis (Figure 5B).

Effect of LL-37 on SAA-induced chemotactic migration in human neutrophils. Isolated human neutrophils (1 × 106 cells/ml of serum free RPMI 1640 medium) were added to the upper wells of a 96-well chemotaxis chamber. Next, the migration across the 3 µm pore size polycarbonate membrane was assessed after incubating at 37℃ for 1.5 h. Various concentrations of LL-37 or SAA were used for the chemotaxis (A). The isolated human neutrophils were incubated in the absence or presence of LL-37 (10 µM) for 10 min prior to the chemotaxis assay using SAA (1 µM or 2 µM) (B). The number of migrated cells was assessed by counting in 5 high power fields (400×). The data are expressed as the means ± SE of three independent experiments performed in duplicate (A, B).
LL-37 does not bind to SAA directly
The finding that LL-37 specifically inhibits SAA-stimulated IL-8 production led us to check the possibility that LL-37 directly binds to SAA, which subsequently results in the inhibition of SAA's action. For this, we performed a BIACore analysis. At first SAA (Figure 6A) or LL-37 (Figure 6C) were immobilized on a CM5 chip. Application of LL-37 to SAA-immobilized chip did not induce positive interaction (Figure 6B). We did not observe positive interaction even in the reverse situation, where SAA was passed through as an analyte to the LL-37 immobilized chip (Figure 6D).

LL-37 does not bind to SAA directly. The ligand (A: SAA, C: LL-37) was immobilized onto CM5 sensor chip. 1 µM of each analyte (B: LL-37 for SAA-immobilized chip, D: SAA for LL-37 immobilized chip) was passed over the chip at the flow rate of 30 µl/min. The binding response was monitored with BIAcore 2000. The results are representative of two independent experiments.
Discussion
We investigated the effect of LL-37 on the SAA-induced IL-8 production and chemotactic migration in human neutrophils. We found that LL-37 inhibited SAA-induced IL-8 production in a concentration-dependent manner. Moreover, this effect of LL-37 on SAA-induced IL-8 production was found to be associated with the inhibition of IL-8 mRNA accumulation, thus suggesting that LL-37 possesses inhibitory properties against SAA-induced IL-8 production at the transcriptional level. We also determined that LL-37 inhibits SAA-stimulated IL-8 production via the downstream activation of FPRL1. LL-37 also inhibited SAA-stimulated neutrophil chemotaxis. Thus, this study provides the first evidence that LL-37 inhibits the action of SAA as a function of IL-8 production and chemotactic migration in human neutrophils.
Previous reports have suggested that SAA has a pro-inflammatory mediator and is highly elevated; both in the circulation and locally in tissues during various pathologic conditions including infection (Jensen and Whitehead, 1998; Uhlar and Whitehead, 1999). Even though the induction of an acute-phase protein such as SAA is necessary for the defense mechanism against infection, the production of abnormally high levels of SAA can cause severe inflammatory responses (Jensen and Whitehead, 1998; Uhlar and Whitehead, 1999). We know that marked increases in SAA have been used as important indicators of the presence of inflammatory diseases (Jensen and Whitehead, 1998; Uhlar and Whitehead, 1999). Keeping in mind that SAA acts as an important mediator of the inflammatory response, researchers have placed more importance on identifying a putative inhibitor of SAA-induced signaling. In this study, we demonstrate that SAA directly stimulates IL-8 production, the chemotactic migration of neutrophils, and an immune-modulating peptide (LL-37) which specifically inhibits SAA-induced IL-8 production and neutrophil chemotaxis. This result indicates that LL-37 can negatively regulate SAA-induced signaling and cellular responses in human neutrophils.
We showed that FPRL1, a chemoattractant receptor, plays a role in the inhibitory effect of LL-37 on SAA-induced IL-8 production. SAA, an endogenous ligand of FPRL1, was found to stimulate IL-8 production in human neutrophils by activating ERK and p38 MAPK. Consequently, it was unclear whether another receptor was involved in SAA-stimulated IL-8 production and the inhibitory effect of LL-37 in this process. However, subsequent to our study, we determined that IL-8 production is induced by SAA in FPRL1-expressing RBL-2H3 cells, but not in vector-transfected RBL-2H3 cells (Figure 4). These results strongly indicate that SAA stimulates the production of IL-8 via FPRL1, whereas LL-37 inhibits SAA-induced IL-8 production by acting on FPRL1. As shown in Figure 6, LL-37 does not directly bind to SAA. From the results we can rule out the possibility that LL-37 directly binds to SAA, resulting in the inhibition of SAA's action. The results also support our notion that LL-37 inhibits SAA-induced cellular signaling and response by acting on FPRL1.
In terms of the intracellular signaling mechanism involved in the LL-37 induced inhibitory effect on SAA-induced cytokine production, we found that LL-37 dramatically blocks SAA-stimulated MAPK activity in human neutrophils (Figure 3B). ERK is essential for the expression of IL-8, which is triggered by the activation of human neutrophils by SAA (Figure 3A). Previous reports have also demonstrated that ERK activity induction by extracellular stimuli is essential for the expression of several pro-inflammatory cytokines such as IL-8. In the present study, we found that SAA-induced p38 MAPK activity was similarly almost completely inhibited by LL-37 in human neutrophils. Taken together, the results indicate that LL-37 inhibits SAA-induced IL-8 production and MAPK activation, which suggests that LL-37 blocks SAA-induced IL-8 production by blocking downstream signaling (MAPK) of FPRL1 induced by SAA.
With regards to the ligand selectivity of FPRL1-mediated IL-8 production, we found that of all the FPRL1 agonists tested (SAA, MMK-1, WKYMVm, and LL-37), only SAA stimulated IL-8 production (Figure 1A). In terms of the mechanisms involved in the differential effects of SAA or WKYMVm on the production of IL-8, we have already demonstrated that SAA does not inhibit [125I]WKYMVm in the binding of FPRL1 (Lee et al., 2005), which suggests that SAA and WKYMVm bind to FPRL1 at different sites. As for the uniqueness of the inhibitory effect of LL-37 on SAA-induced IL-8 production in human neutrophils, this differential effect of FPRL1 ligands on IL-8 production by SAA may also be mediated by the overalapping or non-overlapping binding of these ligands to FPRL1.
In conclusion, we demonstrated for the first time that LL-37 inhibits SAA-induced IL-8 production via FPRL1 in human neutrophils. This inhibitory effect of LL-37 is achieved through the inhibition of ERK- and p38 MAPK-dependent pathways. LL-37 was also found to inhibit the SAA-induced chemotactic migration of human neutrophils. Our results suggest that the action of LL-37 on FPRL1 will be one of the useful negative regulators of SAA's action. Consequently, LL-37 may be regarded as a new anti-inflammatory molecule as well as being potentially considered as a therapeutic agent against inflammatory disorders.
Methods
Materials
Recombinant human SAA (endotoxin level <0.1 ng/µg) was purchased from Peprotech (Rocky Hill, NJ). LL-37 (Yang et al., 2000), MMK-1 (LESIFRSLLFRVM) (Klein et al., 1998), WKYMVm (Seo et al., 1997), and acetylated F2L (Ac-MLGMIKNSLFGSVETWPWQVL) (Migeotte et al., 2005) were synthesized from Anygen Co., Ltd. (Gwang-ju, Korea). FBS and RPMI 1640 medium were purchased from Invitrogen Corp. (Carlsbad, CA). Histopaque-1077, fMLF, L-glutamine, antibiotic-antimycotic solution (10,000 UI/ml penicillin, 10 µmg/ml streptomycin, and 25 µg/ml amphotericin B, were purchased from Sigma (St. Louis, MO). 2'-amino-3'-methoxyflavone (PD98059) and 4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1H-imi dazole (SB203580) were from Calbiochem (San Diego, CA). Rabbit anti-human antibodies to ERK, phospho-ERKs, and phospho-p38 MAPK from Cell Signaling Technology, Inc. (Beverly, MA). HRP-conjugated antibodies to rabbit IgG were purchased from Kirkegaard & Perry, Inc. (Gaithersburg, MD).
Isolation of human neutrophils
Peripheral blood was collected from healthy donors, and human neutrophils were isolated by dextran sedimentation, hypotonic lysis of erythrocytes, and with a lymphocyte separation medium gradient, as described previously (Bae et al., 2001). The isolated neutrophils were maintained in the RPMI 1640 medium, which was supplemented with 10% FBS at 37℃, and was promptly used.
Cell culture
Rat basophilic leukemia cell line RBL-2H3 expressing human FPRL1 was described previously and maintained in DMEM supplemented with 20% FBS at about 1 × 106 cells/ml under standard incubator conditions (humidified atmosphere, 95% air, 5% CO2, 37℃) (He et al., 2000; Bae et al., 2003).
Cytokine assay
Cytokine measurement was performed as previously described (Jo et al., 2004). Neutrophils (3 × 106 cells/0.3 ml) were placed in RPMI 1640 medium containing 5% FBS in 24-well plates and kept in a 5% CO2 incubator at 37℃. After stimulation, cell-free supernatants were collected, centrifuged, and measured for IL-8 by enzyme-linked immunosorbent assay (BD Biosciences Pharmingen, San Diego, CA) according to the instruction of the vender.
Stimulation of cells with SAA for Western blot analysis
Human neutrophils (2 × 106) were stimulated with the indicated concentrations of SAA for predetermined times. After stimulation, the cells were washed with serum free RPMI 1640 medium and lysed in lysis buffer (20 mM Hepes, pH 7.2, 10% glycerol, 150 mM NaCl, 1% Triton X-100, 50 mM NaF, 1 mM Na3VO4, 10 µg/ml leupeptin, 10 µg/ml aprotinin, and 1 mM PMSF). Detergent insoluble materials were pelleted by centrifugation (12,000 × g, 15 min, at 4℃), and the soluble supernatant fraction was removed and stored at either -80℃ or used immediately. Protein concentrations in the lysates were determined using Bradford protein assay reagent.
Electrophoresis and Western blot analysis
Proteins were separated in 10% SDS-polyacrylamide gel and, the proteins were blotted onto a nitrocellulose membrane, which was then blocked by incubating with TBST (Tris-buffered saline, 0.05% Tween-20) containing 5% non-fat dry milk. Subsequently, membranes were incubated with specific antibodies and washed with TBST. Antigen-antibody complexes were visualized after incubating the membrane with 1:5000 diluted goat anti-rabbit IgG antibody coupled to horseradish peroxidase and detected by ECL.
RT-PCR analysis
Human neutrophils, vector- or FPRL1-expressing RBL-2H3 cells (1 × 106 cells) were stimulated with 2 µM SAA in a total volume of 0.2 ml for several lengths of time. The primers used for the RT-PCR analysis have been reported previously (Ushigoe et al., 2000; Jo et al., 2004). The sequences of the primer used were as follows; human IL-8: sense, 5'-ATGACTTCCAAGCTGGCCG-3'; anti-sense, 5'-CTCAGCCCTCTTCAAAAACTT-3'. Human GAPDH: sense, 5'-GATGACATCAAGAAGGTGGTGAA-3', anti-sense, 5'-GTCTTACTCCTTGGAGGCCATG T-3'. Rat IL-8: sense, 5'-ATGGTCTCAGCCACCCGCTCG-3'; anti-sense, 5'-ACTTGGGGACACCCTTTAGC-3'. Rat actin: sense, 5'-ATGGATGATGATATCGCCGCG-3'; anti-sense, 5'-TCTCCATGTCGTCCCAGTTG-3'. We ran 30 PCR cycles at 94℃ (denaturation, 1 min), 62℃ (annealing, 1 min), and 72℃ (extension, 1 min). PCR products were electrophoresed on a 2% agarose gel and visualized by ethidium bromide staining.
Chemotaxis assay
Chemotaxis assays were performed using multiwell chambers (Neuroprobe Inc. Gaithersburg, MD) as previously described (Bae et al., 2001). Briefly, prepared human neutrophils were suspended in RPMI at 1 × 106 cells/ml, and 25 µl of this suspension was placed into the upper well of a chamber separated by a 3 µm polyhydrocarbon filter from the peptide containing lower well. Migrated cells were stained with hematoxylin and then counted in five randomly chosen high power fields (400×) (Bae et al., 2001).
BIAcore assay
Interaction of LL-37 with SAA was analyzed by surface plasmon resonance spectroscopy according to a previous report (Yang et al., 2005). HEPES-buffered saline buffer (10 mM HEPES pH 7.4, 150 mM NaCl) was used as running buffer in all BIAcore experiments. LL-37 or SAA were immobilized on a CM5 sensor chip (BIAcore AB, Uppsala, Sweden) via amine coupling by carbomide chemistry using an amine coupling kit (BIAcore AB, Uppsala, Sweden). Analyte samples (SAA or LL-37) were applied to bind to the immobilized LL-37 or the immobilized SAA, respectively at a fixed flow rate (30 µl/min). The binding was monitored with BIAcore 2000 (BIAcore AB, Uppsala, Sweden).
Data analysis
The results are expressed as means ± SE of the number of determinations indicated. The student's t test was used to compare individual treatments with their respective control values. Significance was accepted when P < 0.05.
Abbreviations
- FPRL1:
-
formyl peptide receptor-like 1
- PD98059:
-
2'-amino-3'-methoxyflavone
- SAA:
-
serum amyloid A
- SB203580:
-
4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1-Himidazole
- WKYMVm:
-
Trp-Lys-Tyr-Met-Val-D-Met-CONH2
References
Badolato R, Wang JM, Murphy WJ, Lloyd AR, Michiel DF, Bausserman LL, Kelvin DJ, Oppenheim JJ . Serum amyloid A is a chemoattractant: induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes . J Exp Med 1994 ; 180 : 203 - 209
Bae YS, Bae H, Kim Y, Lee TG, Suh PG, Ryu SH . Identification of novel chemoattractant peptides for human leukocytes . Blood 2001 ; 97 : 2854 - 2862
Bae YS, Yi HJ, Lee HY, Jo EJ, Kim JI, Lee TG, Ye RD, Kwak JY, Ryu SH . Differential activation of formyl peptide receptor-like 1 by peptide ligands . J Immunol 2003 ; 71 : 6807 - 6813
Chiang N, Fierro IM, Gronert K, Serhan CN . Activation of lipoxin A(4) receptors by aspirin-triggered lipoxins and select peptides evokes ligand-specific responses in inflammation . J Exp Med 2000 ; 191 : 1197 - 1208
Coxon PY, Rane MJ, Uriarte S, Powell DW, Singh S, Butt W, Chen Q, McLeish KR . MAPK-activated protein kinase-2 participates in p38 MAPK-dependent and ERK-dependent functions in human neutrophils . Cell Signal 2003 ; 15 : 993 - 1001
Deng X, Ueda H, Su SB, Gong W, Dunlop NM, Gao JL, Murphy PM, Wang JM . A synthetic peptide derived from human immunodeficiency virus type 1 gp120 downregulates the expression and function of chemokine receptors CCR5 and CXCR4 in monocytes by activating the 7-transmembrane G-protein-coupled receptor FPRL1/LXA4R . Blood 1999 ; 94 : 1165 - 1173
He R, Tan L, Browning DD, Wang JM, Ye RD . The synthetic peptide Trp-Lys-Tyr-Met-Val-D-Met is a potent chemotactic agonist for mouse formyl peptide receptor . J Immunol 2000 ; 165 : 4598 - 4605
He R, Sang H, Ye RD . Serum amyloid A induces IL-8 secretion through a G protein-coupled receptor, FPRL1/LXA4R . Blood 2003 ; 101 : 1572 - 1581
Jensen LE, Whitehead AS . Regulation of serum amyloid A protein expression during the acute-phase response . Biochem J 1998 ; 334 : 489 - 503
Jo EJ, Lee HY, Lee YN, Kim JI, Kang HK, Park DW, Baek SH, Kwak JY, Bae YS . Group IB secretory phospholipase A2 stimulates CXC chemokine ligand 8 production via ERK and NF-kappa B in human neutrophils . J Immunol 2004 ; 173 : 6433 - 6439
Johnson GL, Lapadat R . Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases . Science 2002 ; 298 : 1911 - 1912
Klein C, Paul JI, Sauve K, Schmidt MM, Arcangeli L, Ransom J, Trueheart J, Manfredi JP, Broach JR, Murphy AJ . Identification of surrogate agonists for the human FPRL-1 receptor by autocrine selection in yeast . Nat Biotechnol 1998 ; 16 : 1334 - 1337
Le Y, Gong W, Li B, Dunlop NM, Shen W, Su SB, Ye RD, Wang JM . Utilization of two seven-transmembrane, G protein-coupled receptors, formyl peptide receptor-like 1 and formyl peptide receptor, by the synthetic hexapeptide WKYMVm for human phagocyte activation . J Immunol 1999 ; 163 : 6777 - 6784
Lee HY, Kim MK, Park KS, Bae YH, Yun J, Park JI, Kwak JY, Bae YS . Serum amyloid A stimulates matrix-metalloproteinase-9 upregulation via formyl peptide receptor like-1-mediated signaling in human monocytic cells . Biochem Biophys Res Commun 2005 ; 330 : 989 - 998
Lee HY, Jo SH, Lee C, Baek SH, Bae YS . Differential production of leukotriene B4 or prostaglandin E2 by WKYMVm or serum amyloid A via formyl peptide receptor-like 1 . Biochem Pharmacol 2006 ; 72 : 860 - 868
Maddox JF, Hachicha M, Takano T, Petasis NA, Fokin VV, Serhan CN . Lipoxin A4 stable analogs are potent mimetics that stimulate human monocytes and THP-1 cells via a G-protein-linked lipoxin A4 receptor . J Biol Chem 1997 ; 272 : 6972 - 6978
Migeotte I, Riboldi E, Franssen JD, Grégoire F, Loison C, Wittamer V, Detheux M, Robberecht P, Costagliola S, Vassart G, Sozzani S, Parmentier M, Communi D . Identification and characterization of an endogenous chemotactic ligand specific for FPRL2 . J Exp Med 2005 ; 201 : 83 - 93
Seo JK, Choi SY, Kim Y, Baek SH, Kim KT, Chae CB, Lambeth JD, Suh PG, Ryu SH . A peptide with unique receptor specificity: stimulation of phosphoinositide hydrolysis and induction of superoxide generation in human neutrophils . J Immunol 1997 ; 158 : 1895 - 1901
Su SB, Gao J, Gong W, Dunlop NM, Murphy PM, Oppenheim JJ, Wang JM . T21/DP107, A synthetic leucine zipper-like domain of the HIV-1 envelope gp41, attracts and activates human phagocytes by using G-protein-coupled formyl peptide receptors . J Immunol 1999a ; 162 : 5924 - 5930
Su SB, Gong W, Gao JL, Shen W, Murphy PM, Oppenheim JJ, Wang JM . A seven-transmembrane, G protein-coupled receptor, FPRL1, mediates the chemotactic activity of serum amyloid A for human phagocytic cells . J Exp Med 1999b ; 189 : 395 - 402
Uhlar CM, Whitehead AS . Serum amyloid A, the major vertebrate acute-phase reactant . Eur J Biochem 1999 ; 265 : 501 - 523
Urieli-Shoval S, Linke RP, Matzner Y . Expression and function of serum amyloid A, a major acute-phase protein, in normal and disease states . Curr Opin Hematol 2000 ; 7 : 64 - 69
Ushigoe K, Irahara M, Fukumochi M, Kamada M, Aono T . Production and regulation of cytokine-induced neutrophil chemoattractant in rat ovulation . Biol Reprod 2000 ; 63 : 121 - 126
Yang D, Chen Q, Schmidt AP, Anderson GM, Wang JM, Wooters J, Oppenheim JJ, Chertov O . LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells . J Exp Med 2000 ; 192 : 1069 - 1074
Yang SP, Kwon BO, Gho YS, Chae CB . Specific interaction of VEGF165 with beta-amyloid, and its protective effect on beta-amyloid-induced neurotoxicity . J Neurochem 2005 ; 93 : 118 - 127
Acknowledgements
This paper was supported by Dong-A University Research Fund in 2007.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lee, H., Kim, S., Shim, J. et al. LL-37 inhibits serum amyloid A-induced IL-8 production in human neutrophils. Exp Mol Med 41, 325–333 (2009). https://doi.org/10.3858/emm.2009.41.5.036
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2009.41.5.036
