
Abstract
Coexistence of different hereditary diseases is a known phenomenon in populations with a high consanguinity rate. The resulting clinical phenotypes are extremely challenging for physicians involved in the care of these patients. Here we describe a 6-year-old boy with co-occurrence of a homozygous splice defect in OSTM1, causing infantile malignant osteopetrosis, and a loss-of-function variant in MANEAL, which has not been associated with human disease so far. The child suffered from severe infantile-onset neurodegeneration that could not be stopped by bone marrow transplantation. Magnetic resonance imaging demonstrated global brain atrophy and showed hypointensities of globus pallidus, corpora mamillaria, and cerebral peduncles, which were comparable to findings in neurodegeneration with brain iron accumulation disorders. LC-MS/MS analysis of urine and cerebrospinal fluid samples revealed a distinct metabolic profile with accumulation of mannose tetrasaccharide molecules, suggestive of an oligosaccharide storage disease. Our results demonstrate that exome sequencing is a very effective tool in dissecting complex neurological diseases. Moreover, we suggest that MANEAL is an interesting candidate gene that should be considered in the context of neurological disorders with brain iron accumulation and/or indications of an oligosaccharide storage disease.
Similar content being viewed by others


Introduction
The underlying causes of neurodegenerative disorders in childhood are manifold and straight-forward candidate gene prioritization is challenging. The era of next-generation sequencing has clearly advanced the diagnostic process for affected individuals. Apart from known disease entities, also many novel genetic disorders have been identified and characterized during the last years. In addition, the relevance of clustering of multiple hereditary diseases in consanguineous families has come to attention.1, 2 Coexistence of multiple, functionally relevant gene variants may cause unique clinical syndromes that are extremely difficult to address via routine diagnostics.
Here, we report on an individual of a consanguineous family that suffered from a complex infantile-onset neurodegenerative disorder, characterized by developmental delay, optic nerve atrophy, dyskinetic movement disorder, and a neurodegeneration with brain iron accumulation (NBIA)-like pattern on brain magnetic resonance imaging (MRI).
Case report
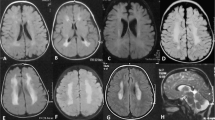
The boy is the third child of healthy, consanguineous parents from Saudi Arabia. Two older brothers and one younger sister are healthy. Pregnancy and birth were reportedly uneventful. First abnormalities were noticed by the parents around the age of 2 months. The infant had feeding difficulties, was irritable and constantly unsettled. Moreover, he did not make eye contact. Further investigations performed in Saudi Arabia led to the identification of a splice defect in OSTM1 consistent with a diagnosis of osteopetrosis. Accordingly, allogeneic stem cell transplantation was successfully performed at the age of 7 months. In the following, the clinical condition of the child stabilized. However, starting between the age of 2 and 3 years, the parents noted regression with loss of previously acquired skills and development of a dyskinetic movement disorder. Further diagnostics performed at different university hospitals including extensive metabolic work-up (including lysosomal enzyme analysis in skin fibroblasts, screening for congenital disorders of glycosylation, etc) and genetic testing (NGS panels for leukodystrophies, peroxisomal disorders, lysosomal storage disorders, etc) did not identify the cause of his neurological deterioration. Brain MRI demonstrated massive brain atrophy (Figure 1a and b) and symmetrical iron accumulation in pallidum (Figure 1c), corpora mamillaria, and cerebral peduncles. Therefore, a disorder within the NBIA spectrum was suspected. At the current age of 6 years, the child is wheelchair-bound and shows severe intellectual disability. Swallowing difficulties required placement of a PEG tube and due to episodic vomiting fundoplication was performed. The boy is unable to communicate and does not make visual contact. Because of his dyskinetic movement disorder various drug regimens were tried; however, without convincing therapeutic effect. Only recently, he suffered from generalized seizures, which were controlled by levetiracetam treatment.

(a) Brain MRI (T1 weighted, coronal view) at the age of 2 years. (b) Brain MRI (T1 weighted, coronal view) at the age of 6 years. Images demonstrate severe brain atrophy during the course of the disease. (c) Susceptibility weighted MRI images (axial view) at the age of 6 years showing bilateral signal alterations within the globus pallidum (white arrows), which are reminiscent of those reported in NBIA disorders. (d) Pedigree of the investigated family and electropherograms showing identified variants in OSTM1 (NM_014028.3, NG_007262.1) and MANEAL (NM_001031740.2). Mutation status of affected (closed symbols) and unaffected (open symbols) family members is depicted. (e) Sashimi plot of RNA sequences produced from patient’s (red, upper panel) and control (blue, lower panel) fibroblast cell lines. These data are in line with the change c.783+5G>T, resulting in a skipping of exon four of the OSTM1 transcript. A full color version of this figure is available at the European Journal of Human Genetics journal online.
Materials and methods
A detailed description of materials and methods used for this paper can be found in the supplementary part of this article.
Results and discussion
On the basis of the complex clinical picture and unusual brain abnormalities whole-exome sequencing was performed. Analysis yielded an average 207-fold coverage of the target region with >98% being covered >20-fold. A search for autosomal homozygous rare (MAF>0.1% in 9000 in-house control exomes) loss of function, splice, and near splice variants prioritized two genes (MANEAL and OSTM1). The splice variant c.783+5G>T, p.(?) in OSTM1 (NM_014028.3, NG_007262.1) was found in a 62 Mb run of homozygosity. In MANEAL (NM_001031740.2), exome sequencing identified the frameshift variant c.446dup, p.(Asp150Argfs*48) located within a 38 Mb homozygous region. Sanger sequencing confirmed the two variants in a homozygous state in the patient with his parents being heterozygous carriers of both variants (Figure 1d). Sequencing of the healthy siblings showed heterozygous changes or wild-type sequences at the respective genomic positions. We next performed whole-transcriptome sequencing on patient-derived fibroblast cell lines to further investigate the functional consequences of the above-mentioned variants. This analysis confirmed that the OSTM1 variant c.783+5G>T results in a complete skipping of exon four of the canonical OSTM1 transcript (Figure 1e). This finding is in line with a loss-of-function character of the c.783+5G>T OSTM1 variant.
The information regarding both variants was submitted to the Leiden Open Variation Database (LOVD v.3.0) (http://databases.lovd.nl/shared/individuals/00100446).
Expression of MANEAL was found to be generally very low in both, patient’s and control fibroblasts (data not shown). Therefore, albeit overall MANEAL expression levels in patient’s RNA were within the lower range of controls this finding is not clearly supportive of nonsense-mediated decay of the mutant transcript. Data from The Human Protein Atlas suggest that MANEAL is mainly expressed in brain tissue (http://www.proteinatlas.org).
OSTM1 encodes osteopetrosis-associated transmembrane protein-1 and functionally relevant variants within this gene may cause infantile malignant osteopetrosis (OMIM #259720), which is a very severe sub-form of this disease. The clinical phenotype includes generalized osteosclerosis, anemia, hepatosplenomegaly, and progressive neurodegeneration. The clinical course is usually fatal during the first years of life. Bone marrow transplantation (BMT) was shown to be ineffective in stopping neurodegeneration and according to current guidelines (00_OP_GUIDELINES_V3.1108015), BMT is contraindicated in patients with OSTM1 defects. Clinical features of the child here include severe optic atrophy and global brain atrophy, which can be attributed to this defect.
However, the MRI abnormalities possibly indicating brain iron accumulation demonstrated in Figure 1c appeared unusual in this context and might be associated with the homozygous frameshift variant in MANEAL. This gene encodes mannosidase endo-alpha-like protein (also known as glycoprotein endo-alpha-1,2-mannosidase-like), which is as a putative membrane protein of the Golgi apparatus, existing as three alternatively spliced isoforms.3, 4 So far, no functional studies regarding MANEAL have been performed. Nevertheless, it was suggested that MANEAL might be involved in glycoprotein metabolism. Interestingly, in several oligosaccharide/glycoprotein storage diseases brain pathology on MRI includes iron accumulation patterns similar to those observed in NBIA disorders.5, 6 The exact pathomechanism behind this phenomenon is unclear. However, lysosomes are known to play a critical role in cellular iron homeostasis.7, 8
In view of these findings, we further investigated a potential role of MANEAL in the disease reported here. Analysis of urine and cerebrospinal fluid samples of the index patient revealed almost identical patterns of diagnostic ions as depicted in Figure 2. We observed a specific elevation of the hexose tetrasaccharides (Hex4 peak at m/z 997). This 'mass spectrum fingerprint' was distinctly different when compared to urine samples of healthy controls or of patients with other lysosomal storage diseases (eg, α-mannosidosis, Pompe disease, and GM1-gangliosidosis; for examples see Supplementary Figure 1) as well as a patient with osteopetrosis caused by disease-causing variants in TCIRG1 (Supplementary Figure 2). Of note, mannose is an epimer of glucose and has the same molecular weight. Therefore, it is not possible to distinguish between them by measuring their mass spectra. To answer the question whether MANEAL deficiency really affects mannose metabolism, we performed enzymatic experiments using recombinant α-mannosidase on patient samples in a time-dependent manner. As depicted in Figure 3, only treatment with α-mannosidase induced a significant decrease of the Hex4 peak up to 50% after 3 h of incubation at 37 °C (Figure 3). In addition, we also performed similar experiments using recombinant α-glucosidase, which revealed no effect on the Hex4 peak (data not shown). Hence we conclude that the Hex4 peak in the MANEAL patient’s body fluids most likely represents mannose moieties.

Tandem mass spectra as parents scan of m/z 175 of the PMP-derivatized control urine sample (a) as well as patient‘s urine (b) and cerebrospinal fluid samples (c). We observed a specific elevation of the hexose tetrasaccharides (Hex4 peak at m/z 997; black arrows) in the patient samples. Images (a, b) depict representative MS/MS spectra of several independent measurements (n=3). The mass ranges between m/z (mass-to-charge) 1160 and 1600 are magnified ten-fold. For additional details regarding the assignments of characteristic diagnostic PMP-derivatized ions to their molecular fragments see Supplementary Table 1.

MS/MS spectra of time-dependent enzymatic digestion experiments using α-mannosidase in patient's urinary oligosaccharide samples. (a–c) As depicted, α-mannosidase treatment induced a time-dependent decrease of the Hex4 peak at m/z 997 (black arrows) indicating that the Hex4 peak represents mannose moieties. The mass ranges betweenm/z (mass-to-charge) 1160 and 1600 are magnified ten-fold.
These findings suggest disturbed mannose metabolism in the affected patient. However, the contribution of this metabolic abnormality to the clinical phenotype is extremely difficult to address. The severe neurological phenotype of OSTM1 deficiency and the effects/side effects of BMT that the patient received at the age of 7 months certainly have a major influence on current clinical problems. Moreover, OSTM1 plays an important role in subcellular endosome/lysosome dispersion and autophagy processes, which could be associated with lysosomal dysfunction.9 Nevertheless, the unusual NBIA pattern on brain MRI, which has not been described for OSTM1 patients, and the above-mentioned findings in body fluids suggest a potential impact of the frameshift variant in MANEAL. Therefore, MANEAL represents certainly an interesting candidate gene that should be considered in the context of neurological disorders with neuroimaging findings suggestive of brain iron accumulation and/or indications of a glycoprotein storage disease.
References
Hmani-Aifa M, Benzina Z, Zulfiqar F et al: Identification of two new mutations in the GPR98 and the PDE6B genes segregating in a Tunisian family. Eur J Hum Genet 2009; 17: 474–482.
Goldenberg-Cohen N, Banin E, Zalzstein Y et al: Genetic heterogeneity and consanguinity lead to a 'double hit': homozygous mutations of MYO7A and PDE6B in a patient with retinitis pigmentosa. Mol Vis 2013; 19: 1565–1571.
Gregory SG, Barlow KF, McLay KE et al: The DNA sequence and biological annotation of human chromosome 1. Nature 2006; 441: 315–321.
Huttlin EL, Ting L, Bruckner RJ et al: The BioPlex Network: a systematic exploration of the human interactome. Cell 2015; 162: 425–440.
Zoons E, de Koning TJ, Abeling NG, Tijssen MA : Neurodegeneration with brain iron accumulation on MRI: an adult case of α-mannosidosis. JIMD Rep 2012; 4: 99–102.
Takenouchi T, Kosaki R, Nakabayashi K, Hata K, Takahashi T, Kosaki K : Paramagnetic signals in the globus pallidus as late radiographic sign of juvenile-onset GM1 gangliosidosis. Pediatr Neurol 2015; 52: 226–229.
Cox TM, Cachón-González MB : The cellular pathology of lysosomal diseases. J Pathol 2012; 226: 241–254.
Bras JM : Lysosomal storage disorders and iron. Int Rev Neurobiol 2013; 110: 251–275.
Pandruvada SN, Beauregard J, Benjannet S et al. Role of Ostm1 cytosolic complex with kinesin 5B in intracellular dispersion and trafficking. Mol Cell Biol 2015; 36: 507–521.
Acknowledgements
TBH was supported by the German Federal Ministry of Education and Research (BMBF) within the framework of the e:Med research and funding concept (grant FKZ 01ZX1405C). EM was supported by the German Research Foundation/Deutsche Forschungsgemeinschaft (KFO217). FD was supported by the German Research Foundation/Deutsche Forschungsgemeinschaft (DI 1731/2-1).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Herebian, D., Alhaddad, B., Seibt, A. et al. Coexisting variants in OSTM1 and MANEAL cause a complex neurodegenerative disorder with NBIA-like brain abnormalities. Eur J Hum Genet 25, 1092–1095 (2017). https://doi.org/10.1038/ejhg.2017.96
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.96
This article is cited by
-
Genetics of Osteopetrosis
Current Osteoporosis Reports (2018)
