
Abstract
For a large number of individuals with intellectual disability (ID), the molecular basis of the disorder is still unknown. However, whole-exome sequencing (WES) is providing more and more insights into the genetic landscape of ID. In the present study, we performed trio-based WES in 311 patients with unsolved ID and additional clinical features, and identified homozygous CPLX1 variants in three patients with ID from two unrelated families. All displayed marked developmental delay and migrating myoclonic epilepsy, and one showed a cerebellar cleft in addition. The encoded protein, complexin 1, is crucially involved in neuronal synaptic regulation, and homozygous Cplx1 knockout mice have the earliest known onset of ataxia seen in a mouse model. Recently, a homozygous truncating variant in CPLX1 was suggested to be causative for migrating epilepsy and structural brain abnormalities. ID was not reported although it cannot be completely ruled out. However, the currently limited knowledge on CPLX1 suggests that loss of complexin 1 function may lead to a complex but variable clinical phenotype, and our findings encourage further investigations of CPLX1 in patients with ID, developmental delay and myoclonic epilepsy to unravel the phenotypic spectrum of carriers of CPLX1 variants.
Similar content being viewed by others

Introduction
CPLX1 encodes complexin 1, a neuronal protein, which belongs to the highly conserved complexin protein family. Presumably, CPLX1 regulates vesicle fusogenicity by interfering with the C-terminal stability of the SNARE complex (soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptor), which is an essential effector of the synaptic vesicle fusion process and the neurotransmitter release cascade.1 Complexin functions at presynaptic nerve terminals to inhibit spontaneous SNARE-mediated synaptic vesicle exocytosis, while enhancing stimulated neurotransmitter relase.2 Variation of CPLX1 is thought to be responsible for loss of its inhibitory function and to enhance spontaneous neurotransmitter release.2 Studies with knockout mice indicate that Cplx1 is implicated in neurological and psychiatric disorders but without ID/developmental delay.3, 4, 5 Recently, Karaca et al. identified in a comprehensive WES study in a large set of consanguineous families with neurodegenerative disorders and brain malformations one homozygous nonsense variant, p.(Glu108Ter) in CPLX1 in two sisters with malignant migrating epilepsy and cortical atrophy. ID was not reported.6
The individuals described here were detected in a whole-exome sequencing (WES) cohort of 311 individuals with unexplained ID (manuscript in preparation). Inclusion criteria to this cohort were developmental delay (DD)/ID with an IQ <70 either as non-syndromic ID (isolated, without additional features, present in 88 out of 311 individuals) or as syndromic ID with additional features (for example craniofacial dysmorphism, organ malformation, aberrant body measurements, etc.) which could not be attributed to a clinically recognizable syndrome by experienced clinical geneticists (in 223 out of 311 individuals). In addition, clinically relevant chromosomal aberrations had to be excluded previously by chromosomal microarray analysis, and fragile-X testing had to be normal. The aim of this study was the analysis of these individuals by WES to provide insight into the genetic basis and to find new candidate genes for ID.
Materials and methods
In this study, we examined 311 patients with unexplained ID. The individuals were clinically assessed by experienced clinical geneticists. Ethics approval was obtained from the respective ethics committees (Ethics vote 5360/13 for the Technische Universität München and 08-3663 for the Universität Duisburg-Essen), and all participants provided written informed consent. The study was conducted in accordance with the Declaration of Helsinki Principles. Trio-based WES was performed as described in Kuechler et al.7 and Sanger sequencing was performed to verify the variants and to perform segregation analysis (Supplementary Material).
Exons are numbered like in the Ensembl 87 entry ENST00000304062.10 (http://www.ensembl.org/Homo_sapiens/Transcript/Summary?db=core;g=ENSG00000168993;r=4:784957-826198;t=ENST00000304062). Variants were deposited at www.LOVD.nl/CPLX1 (patients ID 95415, 95416, 95418).
Results
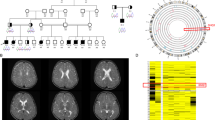
Patient 1 and patient 2 are sisters, the third and fifth children of healthy consanguineous parents originating from Lebanon. The parents are first-degree cousins.
Patient 1 was born in 2005 spontaneously after an uneventful pregnancy at week 41 of gestation. Birth measurements were normal (length: 53 cm (+0.36 SD), weight: 3550 g (−0.07 SD), OFC: 37.5 cm (+1.77 SD)). The first clinical signs of the disease appeared when she was 6 weeks old. She first experienced epileptic spasms. Next, she developed striking seizure exacerbations and migrating myoclonic epilepsy. Seizures occurred more than 100 times daily. Beyond the progressive myoclonic seizures, the diagnosis of generalized epilepsy was established at the age of three years. Seizures were pharmacoresistant and did not respond to ketogenic diet. Electroencephalography (EEG) repeatedly showed generalized epileptiform activity and hyperexcitability. MRI of the brain performed at age 12, 24 and 36 months showed a small cleft of lobule VIII of the left cerebellar hemisphere with malorientation of the adjacent cerebellar folia (Figure 1). No other pathologic findings were noted, especially no cortical atrophy. She was unable to sit unsupported, to crawl or to walk. No head control was achieved. Speech was absent. Assessment at age 25/12 years showed massive hypotonia, normal measurements (height: 88 cm (−0.82 SD), weight: 11 kg (−1.32 SD), OFC: 47.5 cm (−1.26 SD)) and mild craniofacial dysmorphisms (Figure 2). She passed away at 8 years of age most likely due to a severe seizure episode during sleep.

MRI of patient 1 at the age of 12 months. Axial Inversion recovery (left and middle) and coronal FLAIR (right) sequences depicting a small cleft of lobule VIII (arrows) of the left cerebellar hemisphere.

Clinical photographs of the patients with homozygous CPLX1 variants. (a–c) Patient 1 at the age of 25/12 years with hypertelorism, bowed eyebrows, flat midface, long philtrum with thin upper lip and full cheeks. Normal left foot. (d–g) Patient 2 at the age of 29/12 years with hypertelorism, bowed eyebrows, flat midface, thin upper lip. Underriding second toe. Normal hands. (h–k) Patient 3 at the age of seven years with marked eyebrows, large nose with bulbous tip, long philtrum with thin upper lip. Underriding third toe. Normal hands.
Patient 2 was born in 2013. Pregnancy and birth at week 40 were uneventful. Birth measurements were normal (length: 51 cm (−0.32 SD), weight: 3490 g (+0.04 SD), OFC: 35 cm (+0.08 SD)). First non-febrile related epileptic spasms occurred at age 2½ months. During course of disease seizure types were changing. At the age of 6 months, the girl presented with predominantly myoclonic epilepsy. These were progressive and occurred more than 150 times per day. Respiratory infections as well as febrile infections were triggers, leading to generalized seizures. Epilepsy did not respond to antiepileptic drugs. EEG showed generalized spikes and waves as well as hyperexcitability. An MRI scan of the brain performed at age 2 months and ultrasonography of the brain at age 2 and 8 months did not detect any abnormalities, especially no cortical atrophy. At physical examination at around 33 months, she was unable to sit unsupported, to crawl, to walk and to speak. OFC (50 cm, +0.61 SD) and height (90 cm, −0.85 SD) were found to be normal, whereas her weight (9.4 kg) diminished relatively (−2.95 SD). Craniofacial dysmorphisms comprising hypertelorism, bowed eyebrows, flat midface and thin upper lip were noted (Figure 2).
WES detected the homozygous nonsense variant chr4:g.786591G>T (GRCh38/hg38); NM_006651.2:c.315C>A; NP_006642.1:p.(Cys105Ter) in Exon 4 of CPLX1 in the affected sisters. The unaffected parents were found to be heterozygous for this variant.
Patient 3 was born in 2005 as second son of healthy distantly consanguineous parents originating from the Turkmen population. Pregnancy with patient 3 was impaired by placental insufficiency, resulting in a preterm delivery at week 31 of gestation. Birth measurements were normal (length: 39 cm (−0.85 SD), weight: 1350 g (−0.85 SD), OFC: 28 cm (−0.79 SD)). Fetal outcome was poor. He displayed infant respiratory distress syndrome, hypotonia and feeding difficulties (no sucking reflex). Beginning from the first months postpartum, EEG revealed marked persistent generalized seizure activity despite the absence of overt clinical seizure episodes. The first seizure event was recognized in a non-febrile context at 2 years. The boy experienced first myoclonic seizures. During course of disease, seizures switched to alternately myoclonic, tonic and generalized seizures, which occurred >50 times a day. Treatment with distinct anticonvulsants showed no improvement of patient’s clinical signs. He experienced cerebral movement disorder and cerebral palsy. An MRI scan of the brain at around 17 months of age was normal and no cortical atrophy was noted. All milestones of motor and speech development were delayed. He was able to sit at 36 months and to walk with support at age 6 years. Speech was limited to nearly absent. On physical examination at age 7 years, weight (18.5 kg, −2.03 SD) was in the normal range, whereas height (110–112 cm, −2.68 SD) and OFC (48.5 cm, −3.11 SD) were decreased. He presented with mild craniofacial dysmorphisms (Figure 2).
WES identified homozygosity for the variant chr4:g.786,524G>T (GRCh38/hg38); NM_006651.2:c.382C>A; NP_006642.1:p.(Leu128Met) in exon 4 of CLPX1 in the patient and heterozygosity in the unaffected parents.
This variant is known as rs371709824, and six heterozygous but no homozygous allele carriers among 33197 individuals are listed in the ExAC database (http://exac.broadinstitute.org). Three in silico tools (SIFT v5.2.2, PolyPhen-2 and MutationTaster) predict this variant as possibly disease causing. The amino acid exchange affects a highly conserved residue of the C-terminal domain (CTD) of complexin.1 The CTD is required to bind complexin to SNARE-mediated synaptic vesicles (SVs) by membrane interaction and is of particular importance for its inhibitory function.2 Thus, we consider the p.(Leu128Met) as the causative variant in patient 3.
Experimental data for WORM COMPLEXIN indicates that two motifs in the C-terminal domain of CPXL1 are involved in vesicle binding: The first motif spanning residues 110–124 mediates a weak interaction, whereas the second motif comprising residues (128–143) mediates a tight interaction. Both motifs combine to localize and gate complexin function on SVs. (data from Snead et al.).2 The Cys105* variant lacks both of these interaction motifs and the Leu128Met variant exhibits a variant of a conserved residue within the second motif responsible for tight binding.
The clinical features of the patients with homozygous CPLX1 variants are summarized in Table 1.
Discussion
Recently, Karaca et al.6 reported two sisters with homozygous loss-of-function variants in CPLX1 who displayed migrating epilepsy and cortical atrophy without reported ID. The present study is the first to identify homozygous CPLX1 variants in patients with severe myoclonic epilepsy and in addition confirmed marked developmental delay and ID. No further variants were identified.
Although one might conclude that ID in our patients is completely dependent on myoclonic encephalopathy,8 we do not believe that this explanation is sufficient as (i) developmental delay/ID in our patients is remarkably severe and (ii) patient 3 showed ID and developmental delay before onset of epilepsy. This rather points to overlap in terms of the genetic etiology of migrating epilepsy and ID and suggests shared pathological pathways. This hypothesis is supported by studies of Cplx1-deficient mice, which showed marked abnormalities in complex behavior, deficits in sensory and motor skills and suffered from seizures.9, 10, 11 However, the potential functional connection remains unclear.
Complexins are small presynaptic proteins that bind to the SNARE complex where they modulate neurotransmitter release. Cplx1 knockout (Cplx1−/−) mice developed a strong early onset ataxia in the absence of cerebellar degeneration.9 However, Kielar and colleagues reported selective volume loss from the thalamus and cerebellum and atrophy of the molecular layer but no neuron loss in Cplx1−/− mice.5
No such findings were noted in our patients or in the patients reported by Karaca et al. However, in contrast to the findings in the mouse model and our findings, Karaca et al.6 reported cortical atrophy in the homozygous CPLX1 variant carriers. Although we cannot completely exclude that cortical atrophy was not detectable at time of investigation in our patients its presence is rather unlikely because patient 1 was followed-up by MRI brain scans during a period of three years and patient 2 did not show abnormalities pointing to specific brain damage at neuropediatric examination after a disease course of nearly three years. In patient 1, a small cleft of the left cerebellar hemisphere was noted. Cerebellar clefts are rare and thought to be of primary disruptive nature, most often caused by prenatal cerebellar hemorrhage.12 In conclusion, the small number of reported individuals with presumably pathogenic CPLX1 variants impedes a clear statement on whether the cortical atrophy in the patients of Karaca et al. and the cerebellar cleft in our patient 1 may either be coincidental findings or rare side features caused by the CPLX1 deficiency.
We analyzed two independent screening cohorts, comprising 25, respectively 96 index cases with ID and epileptic encephalopathy, respectively spasms, and pedigrees compatible with autosomal-recessive inheritance (Supplementary Material). Among these cases, we did not detect CPLX1 variants or any other pathogenic/likely pathogenic variants or variants of unknown significance in other genes. Due to the rarity of the disorder and the widely dispersed, but very small, patient base no further screening cohort was at our disposal. Nevertheless, our results point to an involvement of CPLX1 in the etiology of both, ID and migrating epilepsy and highlight the need of investigation of CPLX1 in additional, enlarged cohorts of patients with either ID, epilepsy, brain structural abnormalities or combined abnormalities to define the clinical spectrum of CPLX1 variant carriers.
A more precise elucidation of the disease pathophysiology would be a groundbreaking achievement for the understanding of complex neuronal processes and would shed further light on the biology of ID.
References
Trimbuch T, Rosenmund C : Should I stop or should I go? The role of complexin in neurotransmitter release. Nat Rev Neurosci 2016; 17: 118–125.
Snead D, Wragg RT, Dittman JS, Eliezer D : Membrane curvature sensing by the C-terminal domain of complexin. Nat Commun 2014; 5: 4955.
Brose N : Altered complexin expression in psychiatric and neurological disorders: cause or consequence? Mol Cells 2008; 25: 7–19.
Drew CJ, Kyd RJ, Morton AJ : Complexin 1 knockout mice exhibit marked deficits in social behaviours but appear to be cognitively normal. Hum Mol Genet 2007; 16: 2288–2305.
Kielar C, Sawiak SJ, Navarro Negredo P, Tse DHY, Morton AJ : Tensor-based morphometry and stereology reveal brain pathology in the complexin1 knockout mouse. PLoS One 2012; 7: e32636.
Karaca E, Harel T, Pehlivan D et al: Genes that affect brain structure and function identified by rare variant analyses of Mendelian neurologic disease. Neuron 2015; 88: 499–513.
Kuechler A, Willemsen MH, Albrecht B et al: De novo mutations in beta-catenin (CTNNB1) appear to be a frequent cause of intellectual disability: expanding the mutational and clinical spectrum. Hum Genet 2014; 134: 97–109.
McTague A, Howell KB, Cross JH, Kurian MA, Scheffer IE : The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol 2015; 15: 304–316.
Glynn D, Drew CJ, Reim K, Brose N, Morton AJ : Profound ataxia in complexin I knockout mice masks a complex phenotype that includes exploratory and habituation deficits. Hum Mol Genet 2005; 14: 2369–2385.
Glynn D, Sizemore RJ, Morton AJ : Early motor development is abnormal in complexin 1 knockout mice. Neurobiol Dis 2007; 25: 483–495.
Chang S, Reim K, Pedersen M, Neher E, Brose N, Taschenberger H : Complexin stabilizes newly primed synaptic vesicles and prevents their premature fusion at the mouse calyx of held synapse. J Neurosci 2015; 35: 8272–8290.
Bosemani T, Poretti A : Cerebellar disruptions and neurodevelopmental disabilities. Semin Fetal Neonatal Med 2016; 21: 339–348.
Acknowledgements
We are grateful to the families for participating in this study. We thank Sabine Kaya and Daniela Falkenstein for excellent technical assistance. This work was supported in part by the German Ministry of Research and Education (grant numbers 01GS08164, 01GS08167, 01GS08163, German Mental Retardation Network/MRNET) as part of the National Genome Research Network.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Redler, S., Strom, T., Wieland, T. et al. Variants in CPLX1 in two families with autosomal-recessive severe infantile myoclonic epilepsy and ID. Eur J Hum Genet 25, 889–893 (2017). https://doi.org/10.1038/ejhg.2017.52
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.52
This article is cited by
-
Mechanism of Antidepressant Action of (2R,6R)-6-Hydroxynorketamine (HNK) and Its Compounds: Insights from Proteomic Analysis
Molecular Neurobiology (2024)
-
Generation of iPSC lines (KAIMRCi003A, KAIMRCi003B) from a Saudi patient with Dravet syndrome carrying homozygous mutation in the CPLX1 gene and heterozygous mutation in SCN9A
Human Cell (2023)
-
Transcriptomic profiling on localized gastric cancer identified CPLX1 as a gene promoting malignant phenotype of gastric cancer and a predictor of recurrence after surgery and subsequent chemotherapy
Journal of Gastroenterology (2022)
-
Implementation of chromosomal microarrays in a cohort of patients with intellectual disability at the Argentinean public health system
Molecular Biology Reports (2020)
