
Abstract
Inherited retinal dystrophies (IRDs) are clinically and genetically highly heterogeneous, making clinical diagnosis difficult. The advances in high-throughput sequencing (ie, panel, exome and genome sequencing) have proven highly effective on defining the molecular basis of these disorders by identifying the underlying variants in the respective gene. Here we report two siblings affected by an IRD phenotype and a novel homozygous c.1691A>G (p.(Asp564Gly)) ATF6 (activating transcription factor 6A) missense substitution identified by whole exome sequencing analysis. The pathogenicity of the variant was confirmed by functional analyses done on patients’ fibroblasts and on recombinant p.(Asp564Gly) protein. The ATF6Asp564Gly/Asp564Gly variant shows impaired production of the ATF6 cleaved transcriptional activator domain in response to endoplasmic reticulum stress. Detailed phenotypic examination revealed extinguished cone responses but also decreased rod responses together with the ability to discriminate some colours suggestive rather for cone-rod dystrophy than achromatopsia.
Similar content being viewed by others


Introduction
Inherited retinal dystrophies (IRDs) constitute a large, genetically heterogeneous group of degenerative disorders of the retina. The various syndromic and nonsyndromic forms of IRD can be caused by mutations in more than 200 genes.1 Very often, patients suffering from different IRD exhibit overlapping clinical features; therefore, it is difficult to establish the right diagnosis only by clinical characterization. Cone-rod dystrophy (CRD) is a genetically heterogeneous IRD that can be inherited in an autosomal dominant, autosomal recessive or X-linked recessive manner. CRD is characterized by progressive loss of cone function, followed by loss of rod photoreceptor function. Clinical findings include decreased visual acuity, loss of sensitivity in the central visual field, colour vision defects and photophobia, sometimes followed by progressive loss of peripheral vision and night blindness.2 Fundus examination and optical coherence tomography (OCT) performed in patients suffering from CRD typically show changes within the fovea, perifoveal atrophy of the outer retina and ‘bull’s eye’ appearance of the retina.3
Achromatopsia (ACHM, rod monochromacy) is an early-onset autosomal recessive RD characterized by severe photophobia, impaired colour discrimination (colour blindness in the complete form and impaired in the incomplete form), pendular nystagmus and severely reduced visual acuity. Electroretinographic recordings (electroretinography, ERGs) show absent or markedly diminished photopic responses, whereas the function of rod photoreceptors is normal.4 ACHM is considered a stationary disorder, as symptoms do not progress; however, recent analysis has shown that it is actually of progressive nature as cone photoreceptors progressively degenerate.5, 6, 7 ACHM is caused by mutations in genes encoding components of phototransduction cascade: CNGA3, CNGB3, GNAT2, PDE6C and PDE6H.4
Recently, ATF6 (activating transcription factor 6A) variants have been identified as a novel cause of ACHM.8, 9, 10 The ATF6 gene (MIM605537) encodes an activating transcription factor 6α, that is a an endoplasmic reticulum (ER) stress-regulated transmembrane protein.11 In response to protein misfolding in ER or other forms of ER stress, ATF6 regulates the unfolded protein response pathway in parallel with two other ER resident transmembrane proteins: IRE1 and PERK.12 Upon stress, ATF6 migrates from the ER to the Golgi apparatus where it is converted from a full-length 90 kDa transmembrane protein form to its active ~60 kDa cytosolic form by the SP1 and SP2 proteases.11 The ATF6 cytosolic fragment, which contains the basic leucine zipper transcriptional activator translocates to the nucleus, where it activates the transcription of its target genes. ATF6 transcriptionally upregulates many ER protein folding enzymes and chaperones including BiP/GRP78 chaperone.11, 13, 14 By this mechanism, the ATF6 protein helps to maintain ER and cellular homeostasis and alleviates ER stress.15 The ATF6 gene variants identified in ACHM patients include missense, nonsense, splice site, deletion and duplication changes found across the entire gene.8, 9, 10, 16
The overlapping clinical symptoms of ACHM and CRD such as nystagmus, photophobia and colour vision defects observed especially in children suffering from IRD make the precise clinical diagnosis difficult to establish and therefore hamper choosing the right genes for targeted-oriented sequence analysis.
Materials and methods
Clinical diagnosis
This study was conducted in accordance with the tenets of the Declaration of Helsinki. Written informed consent was obtained from the parents as both affected patients were underaged.
The object of the study were two siblings: an 9-year-old boy and his 6-year-old sister of Polish origin with reduced visual acuity and severe photophobia, who were referred for genetic counselling and diagnostics to the ophthalmogenetic clinic (Center for Medical Genetics, Genesis, Poznan, Poland). The family history was taken and the pedigree was carefully checked. The children were earlier referred for full ophthalmological examination, including visual acuity, anterior and posterior segment assessment, full-field ERG and OCT of the macula. Both children were also examined with colour vision test (Ishihara plates); the boy at the age of 8 years and his sister at the age of 5 years. Full-field ERG was performed twice in the boy (at the age of 5 and 9 years) and once in his sister at the age of 6 years. The electrophysiological examination (Reti-port, Roland Consult, Branderburg, Germany) included a flash full-field ERG protocol with the standard scotopic 20 min dark adaptation (rod ERG, standard combined ERG and oscillatory potentials) and 10 min light adaptation (single flash cone ERG, 30 Hz flicker ERG) according to International Society for Electrophysiology of Vision standards.17 The DTL silver treads were used as active electrodes and the gold cup skin electrodes were used as reference and ground ones.
OCT test was done in both affected siblings with SOCT Copernicus device (Optopol Technology, Zawiercie, Poland).
Molecular analysis
DNA samples collection
Blood samples from the affected children as well as their healthy parents were obtained for genetic examination with written informed consent from the parents. Genomic DNA was extracted from peripheral blood using the conventional salting-out procedure.
Nucleotide variants screening with Sanger sequencing and ABCA4 gene SNP microarray
The affected boy’s DNA was firstly screened for known causes of autosomal recessive CRD using a combined strategy of SNP microarray analysis and Sanger sequencing of three genes known to be associated with autosomal recessive CRD (CERKL, ADAM9 and RPGRIP). The primer sequences are available upon a request. PCR products were directly sequenced using the Big Dye Terminator Cycle-Sequencing v3.1 Kit (Applied Biosystems, Foster City, CA, USA) and run on an automated capillary sequencer ABI 3130XL Genetic Analyzer (Applied Biosystems). The nucleotide sequences were analysed with sequence trace alignment software (CodonCode Aligner, CodonCode Corporation, Centerville, MA, USA). In addition, SNP microarray analysis (APEX-array primer extension-based test) of the ABCA4 gene was performed to search for known nucleotide variants (Asper Biotech, Tartu, Estonia).
NGS retinal genes panel
Finally, next generation panel sequencing for eye diseases (CeGaT—Center for Genomics and Transcriptomics, Tuebingen, Germany) was performed in the boy in a scientific set-up. A capture panel of 105 retinal disease-associated genes (IRD panel) was analysed. Details of panel design, library preparation and capture sequencing have already been published.18
Whole exome sequencing
Whole exome sequencing (WES) was performed in the affected girl. WES was conducted with 50 ng of genomic DNA using the Nextera Rapid Capture Exome Kit (Illumina, San Diego, CA, USA) and paired-end sequenced (2 × 100 bp) on Illumina HiSeq1500. The examined sample was sequenced to the mean depth of 51 ×, 87% of target was covered at least 20 × and 96% at least 10 ×. Sequencing data were analysed according to the previously described procedure19 in agreement with the Broad Institute recommendations (https://software.broadinstitute.org/gatk/best-practices/). In brief, sequence readouts were initially analysed with bcl2fastq software to generate reads in fastq format. After the quality control step, including adapter trimming and low-quality reads removal, reads were aligned to the hg19 reference genome with Burrows-Wheeler Alignment Tool (http://bio-bwa.sourceforge.net/) and processed further by Picard and Genome Analysis Toolkit (http://broadinstitute.github.io/picard/)—in particular, base quality score recalibration, indel realignment and duplicate removal were executed, and the single-nucleotide variants (SNVs) and INDELs discovery was performed. Identified variants were annotated with functional information, frequency in population (including ExAC, dbSNP, dbNSFP, 1000 Genomes, as well as the frequency from in-house database of>600 Polish WES), and known association with clinical phenotypes (based on both ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and HGMD (http://www.hgmd.cf.ac.uk/ac/index.php)). The potentially pathogenic novel variant identified with the exome sequencing was validated with Sanger sequencing and tested for co-segregation in both affected children and their healthy parents. The ATF6 gene fragment encompassing the candidate variant was amplified with PCR using the primer pair: 5′-tcctgatccttgggctttgc-3′; 5′-acagcatggctatcaggattaca-3′. The following PCR conditions were applied: 95 °C, 3 min (preliminary denaturation); 40 cycles of denaturation at 94 °C for 30 s, annealing for 30 s with temperature starting from 63 °C, decreasing to 55 °C (touchdown PCR −0.2 °C per cycle), elongation at 72 °C for 45 s and the final synthesis at 72 °C, 10 min. The identified variant was submitted to LOVD database (http://www.lovd.nl/) (patients’ IDs: 00092350 and 00100409).
Cell culture
The skin specimens from dermal biopsy were transferred into a sterile container with Dulbecco’s modified Eagle medium (Gibco, ThermoFisher, Waltham, MA, USA) and immediately delivered to the laboratory (at room temperature). Skin specimens were washed in phosphate-buffered saline and then cut in small pieces and seeded in culture siz-well plates with DMEM medium (Gibco) supplemented with 20% fetal bovine serum (Biochrom, Cambridge, UK) and 1% penicillin/streptomycin (Gibco), and incubated at 37 °C at 5% CO2 to generate explants cultures. Fibroblasts began to migrate out from the explants after a few days. Then the fibroblast cells were passaged to the culture flasks. The medium was exchanged 2 times a week.
HEK293 cells were maintained at 37 °C, 5% CO2 supplemented with 10% fetal calf serum (Mediatech, Manassas, VA, USA) and 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA, USA). To induce ER stress, tunicamycin (TM; Calbiochem EMD Bioscience Inc, Darmstadt, Germany) were dissolved in dimethylformamide and dithiothreitol (DTT) (BioPioneer, Inc., San Diego, CA, USA) was dissolved in water and added to the cell culture media at the indicated concentration.
Plasmid construction and transfection
To generate p.(Asp564Gly) expression plasmids, the variant was introduced into full length ATF6.pcDNA3.1 plasmid by site-direct mutagenesis using overlapping PCR primers. To express wild-type or mutant ATF6 in the cell culture system, plasmids containing the cDNA of this gene were transiently transfected to HEK293 cells using Lipofectamine 2000 (Invitrogen).
Immunoblotting analysis
Human fibroblasts or HEK293 cells expressing wild-type or mutant ATF6 were lysed in SDS lysis buffer (2% SDS, 62.5 mM Tris-HCl pH 6.8, containing protease inhibitors) (Sigma-Aldrich, St Louis, MO, USA), and phosphatase inhibitor (Thermo Scientific, Rockford, IL, USA). Protein concentrations of the total cell lysates were determined by BCA protein assay (Pierce, Rockford, IL, USA). Equal amounts of protein were loaded onto 10% or 4–15% Mini-PROTEAN TGX precast gels (Bio-Rad, Hercules, CA, USA) and analysed by western blot. The following antibodies and dilutions were used: anti-FLAG at 1:5000 (Sigma-Aldrich); anti-human ATF6α antibody at 1:1,000 (Abcam, Cambridge, MA, USA); anti-phosph-eIF2α at 1:1000 (Cell Signaling, Danvers, MA, USA); anti-BiP/GRP78 at 1:1000, anti-GAPDH at 1:5000, anti-HSP90 at 1:5000 and anti-β-tubulin at 1:5000 (GeneTex, Inc., Irvine, CA, USA). After overnight incubation with primary antibody, membranes were washed in TBS with 0.1% Tween-20 followed by incubation of a horseradish peroxidase-coupled secondary antibody (Cell Signaling). Immunoreactivity was detected using the SuperSignal West chemiluminescent substrate (Pierce). Specific western blotting bands were quantified using Bio-Rad Image Lab software. Quantification results are presented as mean±SD from at least three independent experiments. Student’s two-tailed t-tests (for paired samples) were performed to determine P-values.
Endoglycosidase H (Endo H; New England Biolabs, Ipswich, MA, USA) digestion was performed on precleared total cell lysate for 1 h at 37 °C in the buffer supplied by the manufacturer. To preclear the cellular debris from the total lysate, cell lysates were spun at 15 000 rpm at 4 °C for 1 h.
Results
Clinical findings
The unrelated parents of the probands have no ophthalmologic problems. Both children were born from full-term normal pregnancy, by natural delivery and with no complications. Shortly after birth, both children showed the setting-sun eye phenomenon, but neurological investigations gave normal results. At the age of 3 months, congenital nystagmus was observed in the girl only. At the age of 6 months both children started to manifest marked photophobia. The children (boy at the age of 4 years and the girl at the age of 1 year) were referred to the ophthalmogenetic clinic to investigate the precise genetic diagnosis and for genetic counselling. The ophthalmological examination showed reduced visual acuity in both affected children. Best-corrected visual acuity ranged from 0.1 to 0.2.
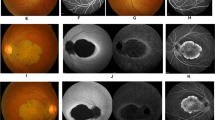
Both patients can recognize majority of colours in everyday life (even such as pink, orange, purple or brown), but the older patient sometimes confuses colours. The colour vision of both patients was more precisely tested using Ishihara colour plates in standard conditions (in the boy at the age of 8 years and in the girl at the age of 5 years). The boy recognized plates number 1, 6–9 and 14–17 easily, and partially plates number 23 (such as patients with deuteranopia) and 24 (such as patients with protanopia). The girl was able to recognize plates number 1, 14–17 (with some difficulties) and partially plates number 2 (such as individuals with red–green deficiency) and 23 (such as patients with deuteranopia). The rest of the plates are not recognizable for both patients. All above mentioned results of the colour vision tests indicate only partial colour vision deficiencies. The full-field flash ERG test performed in the boy at the age of 5 years revealed that the cone responses were totally extinguished and the rod responses were decreased. The ERG examination was repeated in the boy at the age of 9 years and this time also the rod responses were extinguished, showing a progressive nature of the ERG changes observed in the patient (Figure 1a). Moreover, the full-field flash ERG test performed in the girl at the age of 6 years revealed totally extinguished photopic responses and reduced scotopic responses (similarly to the results observed in the boy at the age of 5 years). The OCT scans of both children’s eyes revealed no visible foveal contours, foveal hypoplasia and missing foveal pit. There was also subfoveal atrophy of the photoreceptor layer (Figure 1b).

Clinical phenotype of the affected boy (a) ERG data: left panel—ERGs of normal healthy (7-year-old individual); right panel—ERGs of the patient: scotopic ERG; the upper graph (rod ERG) shows the isolated rods response, which is decreased to the normal value (data in brackets), a-wave 7 μV (normal value: 133 μV), b-wave 54 μV (253 μV); the bottom graph (standard combined ERG) presents rod-cone (combined) responses, a-wave 55 μV (243 μV), b-wave 89 μV (393 μV); oscillatory potentials OP1 25 μV (60 μV), OP2 8 μV (48 μV); photopic ERG: single flash cone ERG and 30 Hz flicker ERG showing non-recordable responses. (b) OCT image showing foveal hypoplasia with missing foveal pit and subfoveal atrophy of the photoreceptors. The small picture at the left side shows the location of the scan. The retinal layers are labelled: IRL, inner retinal layer; IS/OS, photoreceptor inner and outer segments; RPE, retinal pigment epithelium.
Genetic findings
SNP microarrays of the ABCA4 gene and Sanger sequencing of the coding and flanking intronic regions of the CERKL, ADAM9 and RPGRIP genes did not reveal any putative disease-causing variants in the male patient.
Panel sequencing for 105 IRD genes also ruled out variants that affect function in the boy.
WES was performed using a DNA sample of the affected girl. The SNV list from WES was filtered manually in MS Access format. Alignments were viewed with Integrative Genomics Viewer.8 and an autosomal recessive inheritance was considered. The analysis revealed one candidate causative variant: a homozygous substitution c.1691A>G in exon 14 of the ATF6 gene. No additional variants in known autosomal recessive IRD genes were identified. The novel change causes the substitution of aspartic acid to glycine at the position 564 of the amino acid (p.(Asp564Gly)). The variant was described according to transcript reference sequence from Ensembl (ENST00000367942.3).
Segregation analysis of this substitution performed in the affected siblings and their healthy parents revealed that the nucleotide change segregates with the disease phenotype according to an autosomal recessive inheritance (Figure 2). Both affected children appeared to be homozygous for the ATF6 gene variant, whereas both parents are heterozygous carriers for the c.1691A>G substitution.

Chromatograms showing the c.1691A>G mutation in the ATF6 gene. Upper panel: sequence of the homozygous healthy individual (wild-type ATF6 allele); middle panel: sequence of the homozygous affected boy; bottom: sequence of unaffected heterozygous father.
Functional studies
The functional analysis on patient fibroblasts expressing p.(Asp564Gly) and recombinant p.(Asp564Gly) protein was performed to investigate how the amino acid change affects ATF6’s molecular mechanism of signalling and activation. The fibroblasts were obtained from both unaffected heterozygous (ATF6Asp564Gly/+) parents, probands 1 and 2, and two homozygous (ATF6Asp564Gly/Asp564Gly) children, probands 3 and 4 (Figure 3a).

Impaired activation of ATF6 in response to ER stress in p.(Asp564Gly) patient fibroblasts. (a) Pedigree of the family. The heterozygous parents (ATF6Asp564Gly/+) were indicated as probands 1 and 2, for the mother and the father, respectively. The homozygous (ATF6Asp564Gly/Asp564Gly) children were indicated as probands 3 and 4. (b) Patient fibroblasts expressing p.(Asp564Gly) were challenged with TM at the indicated concentration for 24 h. Endogenous ATF6 protein levels were detected by immunoblotting. The level of phosphorylated-eIF2α and BiP/GRP78 were detected by immunoblotting. GAPDH levels were assessed as a loading control. Levels of the BiP/GRP78 protein in patient fibroblasts with or without TM treatment were quantified from independent experiments and are shown after normalization to corresponding GAPDH protein levels. The BiP/GRP78 protein level of patient 1 was set as 1 ×. (c) Patient fibroblasts expressing p.(Asp564Gly) were challenged with DTT for 30 min and ATF6 protein levels were detected by immunoblotting using anti-ATF6 antibody. The middle blot was an over-exposed blot to show the cleaved ATF6 protein. A nonspecific band is indicated with *. HSP90 levels were assessed as a loading control. Levels of the cleaved ATF6 protein in patient fibroblasts with or without DTT treatment were quantified from three independent experiments and are shown after normalization to corresponding HSP90 protein levels. The cleaved ATF6 level of untreated patient 1 was set as 1 ×. Student’s t-test was performed to determine statistical significance.
To induce stress experimentally, proband fibroblasts expressing p.(Asp564Gly) were incubated with TM at the concentration of 5 μg/ml for 24 h. Endogenous ATF6 protein levels and the level of phosphorylated-eIF2α and BiP/GRP78 chaperone proteins were detected by immunoblotting. The levels of full length ATF6 protein in heterozygous and homozygous fibroblasts were equivalent under standard cell culturing conditions (Figure 3b). In response to ER stress induced by TM application, the full-length ATF6 protein levels were upregulated in both heterozygous and homozygous fibroblasts compared with untreated samples (Figure 3b). However, the levels of BiP/Grp78 chaperone protein, a downstream target upregulated when ATF6 is activated by stress, in ATF6Asp564Gly/Asp564Gly fibroblasts were reduced compared with heterozygous controls under experimentally induced stress (Figure 3b). This finding demonstrated that ATF6 signalling was compromised and failed to upregulate its target, BiP/Grp78, in ATF6Asp564Gly/Asp564Gly fibroblasts compared with heterozygous controls. By contrast, phosphorylated eIF2a, a marker for PERK activation, was increased uniformly in both heterozygous and homozygous fibroblasts in response to TM (Figure 3b). Wild-type full-length ATF6 protein undergoes proteolytic cleavage to generate a smaller ~60 kDa cytosolic transcriptional activator fragment in response to ER stress. When we challenged heterozygous and homozygous patient fibroblasts with the ER stress agent, DTT, we observed significantly reduced levels of the cleaved ATF6 cytosolic fragment in homozygous ATF6Asp564Gly/Asp564Gly fibroblasts compared with heterozygous parents (Figure 3c). Taken together, these experiments revealed that the p.(Asp564Gly) variant resulted in the impairment of ATF6 signalling in ATF6Asp564Gly/Asp564Gly fibroblasts through loss of the cleaved cytosolic transcriptional activator fragment of ATF6 upon ER stress.
To ensure that the differences in p.(Asp564Gly) cleaved protein level were not due to fibroblasts cell lines differences, we expressed recombinant FLAG-tagged wild-type ATF6 and mutant p.(Asp564Gly) in HEK293 cells and then challenged with DTT for 1 h. Identical levels of full-length wild-type and variant ATF6 proteins were produced (Figure 4a). However, significantly reduced levels of the cleaved active fragment of the p.(Asp564Gly) protein was observed compared with wild-type ATF6 (Figure 4a), similar to the findings seen in the homozygous ATF6Asp564Gly/Asp564Gly patient fibroblasts (compare Figure 4a with Figure 3c). Production of the cleaved cytosolic fragment of ATF6 from the full-length ATF6 protein requires trafficking of the full-length protein from the ER to the Golgi upon ER stress.15 Full-length ATF6 is a glycoprotein whose sugar tree is trimmed upon translocation from ER to Golgi. Prior studies had shown that an endoglycosidase (Endo) H-resistant isoform of full-length ATF6 was generated when ATF6 had migrated from the ER to Golgi upon ER stress, consistent with the processing of the full-length ATF6’s glycosylated residues. When we compared the Endo-H sensitivity of the wild-type and variant p.(Asp564Gly) proteins, we saw the appearance of an Endo H-resistant full-length ATF6 isoform upon ER stress for the wild-type protein but not with the p.(Asp564Gly) mutant (Figure 4b). This finding revealed that the p.(Asp564Gly) variant failed to generate an Endo H-resistant species consistent with impaired ER to Golgi trafficking upon ER stress. In summary, these findings suggest that the underlying mechanism for loss of signalling seen in the p.(Asp564Gly) variant was the failure of the mutant protein to efficiently traffic from ER to Golgi in response to ER stress, thereby leading to failure to produce the cleaved transcriptional activator fragment.

Impaired ER to Golgi transit of p.(Asp564Gly), during ER stress. (a) Recombinant FLAG-tagged wild-type ATF6 and p.(Asp564Gly) proteins were expressed in HEK293 cells and then challenged with DTT for 1 h. ATF6 protein levels were detected by immunoblotting with anti-FLAG. The band above 95 kDa and the band at 72 kDa represent the full-length FLAG-tagged ATF6 and cleaved FLAG-tagged ATF6, respectively. GAPDH levels were assessed as a loading control. The amount of cleaved ATF6 protein generated from WT-ATF6 and p.(Asp564Gly) was quantified from three independent experiments and shown after normalization to corresponding GAPDH protein levels. The level of WT-ATF6 was set as 100%. (b) Recombinant FLAG-tagged wild-type ATF6 and p.(Asp564Gly) were expressed in HEK293 cells for 20 h and then treated with 2 mM DTT for 90 min. Cell lysates were treated with Endo H glycosidase as indicated. ATF6 protein levels were detected by immunoblotting with anti-FLAG. The Endo H sensitive and insensitive ATF6 bands are indicated. The amount of endo H-resistant ATF6 protein generated from WT-ATF6 and p.(Asp564Gly) was quantified from three independent experiments and are shown after normalization to corresponding GAPDH protein levels. The level of WT-ATF6 was set as 100%. (c) Amino acid sequence alignment of ATF6 proteins from different vertebrate species reveals the perfect conservation of the Asp564 residue.
Discussion
ATF6 mutations constitute a rare cause of IRD. To date, altogether 11 ATF6 mutations have been described.8, 9, 10 Most of the reported patients showed clinical symptoms typical for ACHM: severely reduced visual acuity, photophobia, nystagmus and colour blindness.8, 9 The patient described by Xu et al10—2-year-old girl was too young to complete a colour vision test, but she presented all the remaining symptoms typical for ACHM.
Nystagmus, photophobia and colour vision defects identified as first symptoms especially in small children allow to suspect both incomplete ACHM and CRD diagnosis. Detailed ophthalmological examination as well as genetic analyses help to establish precise diagnosis.
The phenotype exhibited by the children with the homozygous p.(Asp564Gly) variant described herein do not appear to be typical for ACHM, even for the incomplete form. The colour vision impairment in these patients is also not very severe, as they are both able to discriminate many colours in their everyday life. Moreover, Ishihara colour vision tests excluded colour blindness.
The most suggestive clinical feature supporting the diagnosis of CRD, rather than ACHM, in our patients is the diminished scotopic response on ERG. Aside from the totally extinguished ERG photopic responses our patients exhibited also reduced or even totally extinguished scotopic responses, whereas in patients suffering from ACHM rods’ function is normal or nearly normal.20, 21 In the previously reported patients with the ATF6 gene mutations,8, 9 the observed scotopic ERG responses were normal; however, Xu et al10 have described impaired mesopic single-flash responses in the 2-year-old girl with early onset photoreceptor degeneration. Mesopic ERG is performed under low but not quite dark lighting conditions, and allow to examine both/combined photopic and scotopic responses. Although it is difficult to compare the results of ERG under non-standard conditions with the ERG results of our patients, in both cases cone and rod functions are abnormal. Moreover, pigmentary changes in macular region of both eyes reported in the patient described by Xu et al10 may also suggest a kind of photoreceptor dystrophy rather than ACHM (similar to our patients).
Despite the reduced scotopic responses in our patients, the girl does not present night blindness, whereas according to the latest childrens’ mother’s observations the boy sometimes stumbles, especially in the evening. The OCT scan of both our patients revealed foveal hypoplasia and missing foveal pit, similar to the findings detected in all patients with ATF6 gene variants reported to date.8, 10 It confirms the thesis that this specific foveal phenotype can be a characteristic feature of the retinal dystrophy caused by ATF6 gene variants.
The amino acid change p.(Asp564Gly) in the ATF6 gene identified in the affected siblings lies within a luminal domain of the protein. The molecular mechanism underlying pathogenicity of the variant c.1691>G (p.(Asp564Gly)) was tested in patient’s fibroblasts and in recombinant variant p.(Asp564Gly). The functional analysis demonstrated that the p.(Asp564Gly) substitution affects ATF6 signalling and activation by impairment of ER to Golgi trafficking of the full-length protein in response to ER stress. This leads to reduced production of the cleaved ATF6 form that constitutes the transcriptional activator domain, responsible for transcriptional activation of protein folding enzymes and ER chaperones.11 Interestingly, we observed increased levels of the full-length ATF6 form in patient fibroblasts carrying the p.(Asp564Gly) substitution despite reduced levels of the cleaved ATF6 form. We speculate that the cells are increasing the levels of the full-length ATF6 protein, to try to compensate for the fall in cleaved ATF6 levels caused by the p.(Asp564Gly) substitution. Analyses of full-length ATF6 protein levels in cells carrying other hypomorphic ATF6 mutations could reveal if increased full-length ATF6 protein levels are uniformly observed when ATF6 signalling is compromised.
The novel substitution p.(Asp564Gly) is located within a highly conserved C-terminal amino acid motif, very close to the previously reported variant c.1699T>A (p.(Tyr567Asn)) (Figure 4c).8 The patient affected by the p.(Tyr567Asn) substitution described by Kohl et al8 exhibited incomplete ACHM manifested by photophobia, partial foveal hypoplasia and relative central scotoma. Recently, the functional analyses performed on patient’s fibroblasts and recombinant variant p.(Tyr567Asn) protein revealed that the c.1699T>A p.(Tyr567Asn) causes impairment of ATF6 signalling during ER stress.16 We previousy described three classes of the ATF6 gene mutations based on the functional consequences of the analysed mutations. The ATF6 gene mutations included in Class 1 cause loss of function due to defective transport from the ER to Golgi and result in induced cell death and damage during ER stress. Chiang et al16 have also discovered that Class 1 mutants show loss of ATF6 function, without IRE1 and PERK signalling impairment. Thus, both the novel c.1691A>G (p.(Asp564Gly)) variant and the previously reported substitution c.1699T>A (p.(Tyr567Asn)) are included into Class 1.16
This finding seems to be important regarding the possible therapeutic strategies for patients with the ATF6 gene Class 1 mutations. Therapies should be focused on improving protein transport from ER to Golgi apparatus. This could enable SP1 and SP2 proteases effective cleaving of the full-length ATF6 protein, so that active cytosolic domain could transcriptionally upregulate its target genes.16
Photoreceptors are very sensitive to ER stress, which can be triggered by many diverse environmental insults.11, 21 Chronic ER stress induces cell death and thus may contribute to the cone dysfunction and vision loss observed in patients with ATF6 mutations.22, 23, 16
Taking into account the fact that the overlapping clinical symptoms of different IRD procure choosing the proper genes for nucleotide sequence analysis, it seems reasonable to add the ATF6 gene to a list of so-called ‘retinal genes’ analysed by NGS methods.
As the segregation analysis of the variant revealed that both parents are heterozygous carriers of this novel substitution, it is highly probable that they are related. We did not confirm this assumption based on the exact pedigree data, but the parents’ families (mother’s grandfather and father’s grandfather) came from small villages located in very close proximity.
Here we report the first case of CRD caused by a homozygous ATF6 mutation. The findings from functional analysis confirmed the pathogenicity of the novel p.(Asp564Gly) variant. The molecular mechanism of the ATF6 signalling impairment for this luminal substitution appeared to be consistent with the recently described mechanism for Class 1 ATF6 mutations (in which the novel variant was classified).
References
Weisschuh N, Mayer AK, Strom TM et al: Mutation detection in patients with retinal dystrophies using targeted next generation sequencing. PLoS ONE 2016; 11: e0145951.
Hamel CP : Cone rod dystrophies. Orphanet J Rare Dis 2007; 1: 2–7.
Szlyk JP, Fishman GA, Alexander KR, Peachey NS, Derlacki DJ : Clinical subtypes of cone-rod dystrophy. Arch Ophthalmol 1993; 111: 781–788.
Kohl S, Jägle H, Wissinger B Achromatopsia. GeneReviews. University of Washington, Seattle, 1993–2016.
Thiadens AA, Somervuo V, van den Born LI et al: Progressive loss of cones in achromatopsia: an imaging study using spectral-domain optical coherence tomography. Invest Ophthalmol Vis Sci 2010; 51: 5952–5957.
Aboshiha J, Dubis AM, Cowing J et al: A prospective longitudinal study of retinal structure and function in achromatopsia. Invest Ophthalmol Vis Sci 2014; 55: 5733–5743.
Sundaram V, Wilde C, Aboshiha J et al: Retinal structure and function in achromatopsia: implications for gene therapy. Ophthalmology 2014; 121: 234–245.
Kohl S, Zobor D, Chiang WC et al: Mutations in the unfolded protein response regulator ATF6 cause the cone dysfunction disorder achromatopsia. Nat Genet 2015; 47: 757–765.
Ansar M, Santos-Cortez RL, Saqib MA et al: Mutation of ATF6 causes autosomal recessive achromatopsia. Hum Genet 2015; 134: 941–950.
Xu M, Gelowani V, Eblimit A et al: ATF6 is mutated in early onset photoreceptor degeneration with macular involvement. Invest Ophthalmol Vis Sci 2015; 56: 3889–3895.
Haze K, Yoshida H, Yanagi H, Yura T, Mori K : Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell 1999; 10: 3787–3799.
Walter P, Ron D : The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011; 334: 1081–1086.
Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R : Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J Biol Chem 2000; 275: 27013–27020.
Shen J, Chen X, Hendershot L, Prywes R : ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell 2002; 3: 99–111.
Nadanaka S, Yoshida H, Kano F, Muraka M, Mori K : Activation of mammalian unfolded protein response is compatible with the quality control system operating in the endoplasmic reticulum. Mol Biol Cell 2004; 15: 2537–2548.
Chiang WC, Chan P, Kohl S et al: Achromatopsia mutations target sequential steps of ATF6 activation. Proc Natl Acad Sci USA 2016; 114: 400–405.
Marmor MF, Holder GE, Seeliger MW, Yamamoto S International Society for Clinical Electrophysiology of Vision: Standard for clinical electroretinography (2004 update). Doc Ophthalmol 2004; 108: 107–114.
Glockle N, Kohl S, Mohr J et al: Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur J Hum Genet 2014; 22: 99–104.
Ploski R, Pollak A, Muller S et al: Does p.Q247X in TRIM63 cause human hypertrophic cardiomyopathy? Circ Res 2014; 114: e2–e5.
Andreasson S, Tornqvist K : Electroretinograms in patients with achromatopsia. Acta Ophthalmol (Copenh) 1991; 69: 711–716.
Khan NW, Wissinger B, Kohl S : Sieving PA. CNGB3 achromatopsia with progressive loss of residual cone function and impaired rod-mediated function. Invest Ophthalmol Vis Sci 2007; 48: 3864–3871.
Alavi MV, Chiang WC, Kroeger H et al: In vivo visualization of endoplasmic reticulum stress in the retina using the ERAI reporter mouse. Invest Ophthalmol Vis Sci 2015; 56: 6961–6970.
Chan P, Stolz J, Kohl S, Chiang WC, Lin JH : Endoplasmic reticulum stress in human photoreceptor diseases. Brain Res 2016; 1648: 538–541.
Acknowledgements
This study was supported by a grant from the Polish Ministry of Science and Higher Education (806/N-NIEMCY/2010/0). We are grateful to the patients for participating in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Skorczyk-Werner, A., Chiang, WC., Wawrocka, A. et al. Autosomal recessive cone-rod dystrophy can be caused by mutations in the ATF6 gene. Eur J Hum Genet 25, 1210–1216 (2017). https://doi.org/10.1038/ejhg.2017.131
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.131
