
Abstract
Keloids result from abnormal proliferative scar formation with scar tissue expanding beyond the margin of the original wound and are mostly found in individuals of sub-Saharan African descent. The etiology of keloids has not been resolved but previous studies suggest that keloids are a genetically heterogeneous disorder. Although possible candidate genes have been suggested by genome-wide association studies using common variants, by upregulation in keloids or their involvement in syndromes that include keloid formation, rare coding variants that contribute to susceptibility in non-syndromic keloid formation have not been previously identified. Through analysis of whole-genome data we mapped a locus to chromosome 8p23.3-p21.3 with a statistically significant maximum multipoint LOD score of 4.48. This finding was followed up using exome sequencing and led to the identification of a c.1202T>C (p.(Leu401Pro)) variant in the N-acylsphingosine amidohydrolase (ASAH1) gene that co-segregates with the keloid phenotype in a large Yoruba family. ASAH1 is an acid ceramidase known to be involved in tumor formation by controlling the ratio of ceramide and sphingosine. ASAH1 is also involved in cell proliferation and inflammation, and may affect the development of keloids via multiple mechanisms. Functional studies need to clarify the role of the ASAH1 variant in wound healing.
Similar content being viewed by others

Introduction
Scar formation is a complex self-limiting process of dermal wound healing. Keloids can develop when regulatory mechanisms for self-limitation of dermal scarring are compromised. They transgress the margin of the original wound in a tumor-like expansion of scar tissue, and keloid growth can persist for several years. Keloids typically develop after stimulation by trauma or infection. After proliferating for months or years, they form a regressing inner zone while maintaining a proliferating inflammatory outer margin.
Keloids pose a significant health issue and are mostly found in populations of sub-Saharan origin, but can occur in all racial and ethnic groups. The incidence has been estimated to be between 0.09% in the Europeans1 and 6% in a West African population.2 It is estimated that up to 3% of all cases in the West African study2 and 28% in a Chinese population3 comprise inherited keloids.
Keloids develop most commonly in young adults and are less common in children before puberty and in adults over 30 years old.3 The etiology and mechanisms that drive the scar tissue proliferation for prolonged periods of time are still unknown. There is evidence that alterations in inflammatory response, immune cell composition and increased susceptibility of keloid fibroblasts to cytokines and hormones as well as changes in matrix remodeling and apoptosis contribute to the maintenance of fibrotic events in keloids.4 Current treatment of keloids is unsatisfactory as a patient may respond to one treatment but not to others and some patients suffer from a high recurrence rate.5 Differences in expressivity, age of onset and response to treatment suggest that keloid formation is a genetically heterogeneous disorder.
Previous genetic studies provide evidence for autosomal dominant, additive or oligogenic inheritance in families.6, 7 Highly variable results of gene expression studies in families6, 8 and in keloid fibroblasts9, 10, 11 also indicate that keloids are due to heterogeneous genetic events.
ASAH1 encodes one out of five known acid ceramidases, which cleaves ceramide from fatty acid to produce sphingosine, and conversely it can catalyze the synthesis of ceramide using fatty acid and sphingosine.12 The balance between ceramide and sphingosine is important in regulating diverse functions; in particular, the ASAH1 protein is involved in apoptosis, cell cycle, differentiation and cell invasion.13 The ASAH1 protein is highly expressed in cancer cell lines and human alveolar macrophages, suggesting a role in proliferative lesions and inflammation.13 Here we used a combination of linkage analysis and exome sequencing in a large Yoruba family from Nigeria to identify a variant that affects function in the gene encoding acid ceramidase or N-acetylsphingosine amidohydrolase (ASAH1; MIM 613468) as the cause for susceptibility to keloids.
Materials and Methods
Family ascertainment
Family members were recruited according to protocols approved by the Institutional Review Board of University of Connecticut Health and the Ethics Committees of the University of Ibadan/University College Hospital and the College of Health Sciences, Ladoke Akintola University of Technology, Osogbo. Informed consent was obtained from all study participants. Family A is of ethnic Yoruba background. Age at onset varied widely, from the first to the seventh decade of life. Because of the age-dependent onset of keloids, only family members who are 21 years of age and older, and do not present with keloids are considered to be unaffected for the purpose of linkage analysis. Subjects were ascertained by research teams led by experienced plastic surgeons, and all keloids were photographically documented. From the medical history and physical examination, no other skin lesions, craniofacial features, limb anomalies, systemic signs and symptoms that suggest syndromic or inherited neurologic disease were detected.
Genetic analysis
Genomic DNA was isolated from peripheral blood (Gentra Puregene Blood Kit, Qiagen, Germantown, MD, USA) or saliva (Oragene•DNA, DNA Genotek, Ottawa, ON, Canada). Genotyping was performed using the Illumina InfiniumLinkage-24 BeadChip (Illumina, San Diego, CA, USA) that includes 5923 single-nucleotide polymorphism (SNP) markers across the genome.
Quality control of the genotype data was performed using PEDCHECK14 to identify Mendelian inconsistencies and MERLIN15 to detect occurrences of double-recombination events over short genetic distances, which most likely occur due to genotyping error. Two-point and multipoint linkage analyses were performed with Superlink.16 The two individuals with hypertrophic or stretched scars but no keloids were included in the analysis as having an unknown affection status (Figures 1 and 2). Linkage analysis was performed using an autosomal dominant mode of inheritance with 90% penetrance, a disease allele frequency of 0.001 and marker allele frequencies for the Yoruba population from HapMap release #28. The Rutgers combined linkage–physical map of the human genome Build 37 version was used to obtain genetic map positions and distances for the multipoint linkage analysis. Haplotypes were reconstructed using SimWalk2 2.91.17

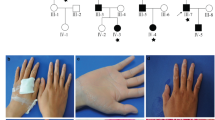
Clinical information for family A. Pictures of keloid and non-keloid scars with physical location of scars for each individual. Each individual is labeled according to the pedigree drawing in Figure 2.

Pedigree of family A and haplotypes within chromosome 8p23.3-p21.3. Proband is indicated by a black arrow. Filled black symbols denote family members with keloids, symbols filled in half-black represent individuals with non-keloidal scars, while clear symbols represent unaffected individuals. Genotypes for each individual are indicated beneath the corresponding symbol and SNP markers are listed at the right upper panel. The haplotype that segregates with keloid occurrence is boxed.
Exome sequencing and variant calling for samples II-3 and II-7 (Figures 1 and 2) was performed at the University of Washington Center for Mendelian Genomics as previously described.18 In brief, sequence capture was performed in solution using the Roche NimbleGen SeqCap EZ Human Exome Library v2.0 (~37 Mb target) (Roche, Indianapolis, IN, USA). Fastq files were aligned to the hg19 human reference sequence using Burrows Wheeler Aligner.19 The variant call format file that was generated using the Genome Analysis Toolkit20 was annotated using ANNOVAR21 and dbNSFP.22 A single-nucleotide variant was selected as potentially causal if the variant is (a) shared by the affected individuals with exome data, (b) falls within the mapped region, (c) has minor allele frequency (MAF) <0.001 in any population within the Exome Aggregation Consortium (ExAC) database and (d) is deemed damaging by at least two bioinformatic tools in dbNSFP. Potentially causal exome variants were tested for co-segregation with keloids by Sanger sequencing using DNA samples from the remaining family members. Sanger sequencing of the ASAH1 c.1202T>C variant was performed using DNA samples from 318 additional Yoruba patients with familial or spontaneous keloid formation and from 192 unaffected Yoruba control individuals who reported no keloids in their families. To examine the possibility of a shared haplotype among unrelated variant carriers, selected exome variants flanking the ASAH1 c.1202T>C variant were Sanger-sequenced in two probands with keloids who were identified to carry the variant. The ASAH1 c.1202T>C variant was assigned the ClinVar ID SCV000538196.
Immunohistochemistry
Normal skin samples were obtained from the National Disease Research Interchange and keloid samples from the University of Ibadan, Nigeria. Samples were embedded in paraffin. Seven-micrometer sections were subjected to antigen retrieval in citrate buffer. Specimen were blocked in 10% BSA and 0.1% Tween 20 before incubation with ASAH1 or control IgG antibodies (Santa Cruz Biotechnology, Dallas, TX, USA). Anti-goat Alexa 594 (Thermo Fisher Scientific, Waltham, MA, USA) was used as secondary antibody. Nuclei were stained with Hoechst 33342 dye (Molecular Probes, Thermo Fisher Scientific, Waltham, MA, USA). Sections were imaged using a Zeiss Observer Z1 microscope (Carl Zeiss Microscopy, Thornwood, NY, USA).
Results
Patient description
Scars in all participating family members were thoroughly examined. Family A consists of 24 informative members, including 9 individuals with keloids and 2 with hypertrophic or stretched scars (Table 1). All unaffected members were older than 21 years at the time of genotyping. The proband (II-2) is a 65-year-old female with multiple large keloids on her torso (Figures 1 and 2). She reported that the first keloid appeared at age 57 years due to acne, and for at least eight other keloids the cause was unknown. Her mother (I-2; Figures 1 and 2) suffers from multiple (≥12) spread-out keloids covering large portions of her torso, which first appeared at age 68 years. The cause for keloids was given as infections. For this individual, keloids were treated with cortisone injections at age 72 years without significant improvement. The proband’s sister (II-7; Figures 1 and 2) suffers from four large keloids on her arms, back and chest. Her first keloids on the upper extremities and chest appeared at 2 years of age after injuries with a sharp object. The keloid on her upper arm appeared at age 44 years, also due to an injury with a sharp object. Another sister of the proband (II-5; Figures 1 and 2) has a single large, non-tender, non-itchy, butterfly-shaped keloid on her abdomen since age 42 years, which was caused by injury with a sharp object. A third sister of the proband (II-3; Figures 1 and 2) developed a small keloid due to bruising on her right leg at age 38 years.
Within the third generation, among the six children of the proband’s sister II-7, four have been diagnosed with keloids (Figures 1 and 2). Niece III-14 has two small itching keloids on her thighs since age 17 due to abrasions. Female III-15 is the twin sister of III-14 and has a small keloid on her calf since age 17 years due to bruising. Also at age 17 she incurred another injury to her back which left a 3 × 2 cm flat scar that did not become a keloid. Because of bruising, nephew III-16 has two small keloids on his legs with onset at age 28 years. Another nephew III-17 has a small keloid on his thigh and a large hypertrophic scar on his arm since age 24 years that were caused by injuries. There was no tenderness or itching at the scar. The youngest siblings III-18 and III-19 were 23 and 18 years old, respectively, at the time of sample ascertainment and have no keloids.
For two family members (III-7 and III-11) the affection status was assigned as unknown (Figures 1 and 2). The proband’s 30-year-old niece III-7 has a 3 × 0.5 cm hypertrophic scar on her knee, therefore she was classified as having unknown affection status. The proband’s niece III-11 (24 years old) is also classified as having unknown affection status because she has a 3 × 1 cm ellipsoid asymptomatic scar on her abdomen that persisted as a raised scar for 2 years before recruitment. No other relevant health issues have been reported for this family.
Linkage analysis
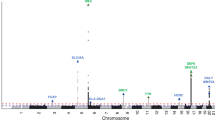
In family A, a statistically significant maximum two-point LOD score of 4.08 (θ=0) was obtained at marker rs2028806 (chromosome 8: 8.93 Mb; Supplementary Table 1). The maximum multipoint LOD score is 4.48 and occurred at three markers rs4840947 (8.23 Mb), rs2028806 (8.93 Mb) and rs1544981 (10.68 Mb). The three-unit support interval (Supplementary Table 1) falls between rs336431 (0.58 Mb) and rs966773 (19.75 Mb). The haplotype that segregates with keloid susceptibility (Figure 2) is flanked by markers rs763869 (1.38 Mb) and rs966773 (19.75 Mb). The haplotype region that segregates with the keloid phenotype is smaller than the three-unit support interval and defines the boundaries of the locus interval. The upper and lower boundaries of the haplotype are due to recombination events that were observed between rs763869 (1.38 Mb) and rs4242539 (1.69 Mb) in individual II-3, and between rs871667 (19.15 Mb) and rs966773 (19.75 Mb) in individual II-2 (Figure 2). The locus interval at chromosome 8p23.3-p21.3 contains 18.37 Mb of sequence, within which are 113 genes. No other genomic region was identified to have a maximum multipoint LOD score >2.0.
Exome and Sanger sequencing
Within the mapped locus interval there were two rare, damaging, nonsynonymous variants that were identified to be shared by the two affected individuals with exome data. They are as follows: (a) AGPAT5 NM_018361.3: c.926G>A (p.(Gly309Glu)) (ClinVar accession SCV000579507), which had a Combined Annotation Dependent Depletion (CADD)23 score of 25.4 and was predicted to be damaging by fathmm,24 the Likelihood Ratio Test (LRT),25 MutationAssessor,26 MutationTaster,27 PROVEAN28 and SIFT;29 and (b) ASAH1 NM_004315.5: c.1202T>C (p.(Leu401Pro); rs368345612), which had a CADD score of 28.1 and was predicted damaging by LRT, MetaLR/MetaSVM from dbNSFP, MutationAssessor, MutationTaster, Polyphen-2,30 PROVEAN and SIFT. Both variants occurred in two heterozygous ExAC African alleles (MAF=0.00019) but were not found in other ExAC populations. Both variants were Sanger-sequenced using DNA samples from the rest of the family members. The AGPAT5 variant was absent in affected participants I-2, II-2, III-7, III-14, III-15, III-16 and III-17 (seven of nine affected individuals) of family A. Only the ASAH1 variant co-segregated with keloids, when reduced penetrance is taken into account. All affected subjects were heterozygous for the ASAH1 variant (Supplementary Figure 1), as well as unaffected individuals III-19 (21 years), III-10 (33 years) and III-12 (30 years). Individual III-11, who had an unusually wide scar that was not diagnosed as keloid, was wild type for the variant. In summary, the ASAH1 variant co-segregated with all affected family members and was also found in young phenotypically unaffected family members III-10 (33 years) and III-12 (30 years), who shared SNP marker haplotypes with affected family members.
In all, 2 out of 318 patients with keloids (MAF=0.003) who were screened for the ASAH1 variant were heterozygous for the c.1202T>C (p.(Leu401Pro)) variant. One patient is a member of a small family B with keloids occurring in three generations, however, only the proband provided a DNA sample. The second patient is a sporadic (non-familial) case. When selected exome variants surrounding the ASAH1 variant were Sanger-sequenced in the two proband carriers with keloids, a 210.4 kb haplotype that includes the ASAH1 c.1202T>C variant was identified to be common among carriers of family A and the two probands with the ASAH1 variant (Supplementary Table 2). The ASAH1 variant was not identified in 192 unaffected Yoruba control individuals who had no family history of keloids.
Immunohistochemistry
ASAH1 staining in keloid scar tissue and normal skin tissue was found in epidermis and dermis (Figure 3). Especially in the dermis of keloid tissue, a number of ASAH1-positive cells were associated with blood vessels. Note, however, that the keloid samples did not derive from patients with the ASAH1 p.(Leu401Pro) variant.

Immunohistochemical staining shows ASAH1 expression in epidermis and associated with blood vessels in keloid scar and normal skin.
Discussion
ASAH1 is ubiquitously expressed, with highest expression in the heart and kidney.31 Increased ASAH1 expression has been reported in the aging kidney,32 while lack of ceramidase activity in the kidney was observed in Farber disease or lipogranulomatosis.33 Farber lipogranulomatosis (MIM 228000) is a lysosomal storage disorder due to autosomal recessive ASAH1 variants34, 35 that leads to accumulation of ceramide in tissues due to enzymatic deficiency, and is primarily characterized by subcutaneous nodules, joint deformities and hoarseness, although severe and atypical forms such as hydrops fetalis, failure to thrive and congenital heart disease have also been reported.36, 37 Autosomal recessive ASAH1 variants can also cause spinal muscular atrophy with progressive myoclonic epilepsy.38 The occurrence of hydrops fetalis in a child who is compound heterozygous for a splice variant and a large deletion mirrors the embryonic lethal phenotype in Asah1−/− mice.39 On the other hand, heterozygous Asah1+/− mice had no overt clinical phenotype, but at 6 months of age, started to develop lipid-laden inclusions in the liver, lung, bone and skin.39 To date no human disease has been associated with heterozygous ASAH1 variants. Individuals heterozygous for variants causing Farber lipogranulomatosis or spinal muscular atrophy with progressive myoclonic epilepsy are not known to have increased risk for keloids.
Here we report an ASAH1 c.1202T>C (p.(Leu401Pro)) variant that, based on an autosomal dominant pattern with reduced penetrance, co-segregates with susceptibility to keloids in a large Nigerian Yoruba family. Interestingly, the same ASAH1 variant was found in two other Nigerian individuals with keloids who also carry the same short haplotype (Supplementary Table 2). ASAH1 does not overlap with keloid loci identified on chromosomes 1, 3 and 15 in a genome-wide association study of two East Asian populations (Japanese and Chinese)40, 41 or with familial loci on 2q23 and 7p11.42 This locus also does not overlap with cytogenetic locations for syndromes or genes that have been previously implicated in keloid pathogenesis, such as Rubinstein–Taybi syndrome (MIM 180849) that is due to heterozygous variants in CREBBP (MIM 600140) at 16p13.3,43 or TGFB1 (MIM 190180) at 19q13.2, which is upregulated in keloid fibroblasts.44 Although the 8p region has not been previously associated with keloids, the statistically significant LOD score that was obtained for family A provides strong evidence that the 8p23.3-p21.3 region contains a keloid gene. Moreover, the lack of additional rare damaging variants across the genome that co-segregate with keloids in family A points to ASAH1 c.1202T>C as the causal variant for keloid susceptibility. As autosomal dominant additive or oligogenic inheritance in families6, 7 cannot be excluded as causative for keloids in this family A, it is possible that another regulatory variant within the 8p23.3-p21.3 locus, which could not be detected by exome sequencing, contributes to keloids in this family A.
Reduced penetrance was used for the analysis due to marked differences in age of onset and degree of scarring even within the same family. In family A, the ages at which the first keloid appeared varied from the first to the seventh decade of life. Individuals with keloids often reported developing more than one keloid around the same age. However, as seen in individuals III-15 and III-17 of family A, it is possible to develop normal scars and keloids at around the same age. Although sustaining a dermal injury is necessary for the development of keloids, we hypothesize that due to the late age of onset for many of the affected family members, additional factors that change over time must have a role, as it is unlikely that they did not incur injuries earlier in life. The late onset of keloids in this family is not typical for most families with inheritable keloids. While the proband II-2 developed her first keloid at age 57 and her mother I-2 even later at age 68, the sister II-7 was reported to express her first keloid at 2 years of age. This variability in onset and expressivity is most likely due to environmental and/or genetic modifiers, such as hormonal status at time of injury, inflammatory status and somatic variants that affect skin in certain parts of the body (eg, skin exposed to UV radiation or chemical injury). In addition, the degree of penetrance may be specific to the gene or variant segregating with keloids in a family. Given the wide age range at which keloids initially developed in this family, individuals who are currently unaffected but carry the ASAH1 variant (individuals III-10, III-12 and III-19) might develop keloids later in life. In this analysis the inclusion of unaffected individuals with a minimum age of 21 years is based on an arbitrarily assigned age cutoff and it is very difficult to assess at which age variant carriers will develop keloids due to the wide variability in age of onset for familial keloids.6 On the other hand, affection status was made unknown for two individuals who have hypertrophic or stretched scars (Figure 2). Individual III-7 who has a hypertrophic scar carries the haplotype that segregates with keloid susceptibility (Figure 2). We believe that hypertrophic scars may be part of the keloid spectrum and that common pathways or genes are involved in the development of either keloid or hypertrophic scars. Despite this variability in penetrance and affection status, the highly statistically significant LOD score that was achieved on chromosome 8p23.3-p21.3 strongly supports the occurrence of a gene for keloid susceptibility within the mapped locus. In addition, within the same locus interval affected-only analysis resulted in a suggestive LOD score of 2.11, which is the maximum LOD score that is achievable for this family in an affected-only analysis.
Immunohistochemistry revealed ASAH1 staining in both normal skin and keloid tissue (Figure 3). ASAH1 is expressed in endothelial cells, in perivascular cells and in keratinocytes (Figure 3), which are cell types that have been suspected to be involved in keloid fibrosis. In previous studies comparing keloid tissue with normal skin, specific ceramides may be lower in keloid tissue45, 46 and failed to be activated as a second messenger in Fas-mediated apoptotic signal transduction.47 Keloid fibroblasts were also more resistant to ceramide-induced apoptosis compared to normal dermal fibroblasts.48 In Asah+/− mice with ceramidase deficiency, ceramide accumulation in the liver and lipid-laden inclusions with lamellar-like structures and crystalline storage material were observed in several tissues, including skin.39 This description seems similar to the tubular inclusions in dermal histiocytes and zebra-body-like and needle-like lysosomal inclusion in epidermal cells of Farber disease patients.49, 50 Furthermore, acid ceramidase levels in cultured skin fibroblasts from heterozygous parents of Farber disease patients were at 30–60% less than those from skin of control subjects.51 Unfortunately, we were unable to obtain keloid tissue from carriers of the ASAH1 c.1202T>C variant, thus the pathologic profile and the ceramide and ceramidase levels in keloids expressing the variant cannot be verified.
In conclusion, we identified ASAH1 as a gene that is involved in keloid susceptibility. Several treatment modalities such as enzyme replacement therapy, gene therapy and hematopoietic stem cell transplantation are currently being explored for treatment of Farber disease, and acid ceramidase is identified as a potential therapeutic target for other diseases as well, for example, bacterial infection, diabetes, arthritis, retinal disease and cancer.52 The identification of an ASAH1 variant as a cause of keloid susceptibility likewise implicates acid ceramidase as a potential target for keloid treatment.
References
Woringer F : in: Nouvelle Pratique Dermatologique. Paris: Masson & Co, 1936; 6: 561.
Omo-Dare P : Genetic studies on keloid. J Natl Med Assoc 1975; 67: 428–432.
Lu WS, Zheng XD, Yao XH, Zhang LF : Clinical and epidemiological analysis of keloids in Chinese patients. Arch Dermatol Res 2015; 307: 109–114.
Shih B, Garside E, McGrouther DA, Bayat A : Molecular dissection of abnormal wound healing processes resulting in keloid disease. Wound Repair Regen 2010; 18: 139–153.
Leventhal D, Furr M, Reiter D : Treatment of keloids and hypertrophic scars: a meta-analysis and review of the literature. Arch Facial Plast Surg 2006; 8: 362–368.
Clark JA, Turner ML, Howard L, Stanescu H, Kleta R, Kopp JB : Description of familial keloids in five pedigrees: evidence for autosomal dominant inheritance and phenotypic heterogeneity. BMC Dermatol 2009; 9: 8.
Marneros AG, Norris JE, Olsen BR, Reichenberger E : Clinical genetics of familial keloids. Arch Dermatol 2001; 137: 1429–1434.
Bayat A, Arscott G, Ollier WE, Ferguson MW, Mc Grouther DA : Description of site-specific morphology of keloid phenotypes in an Afrocaribbean population. Br J Plast Surg 2004; 57: 122–133.
Satish L, Lyons-Weiler J, Hebda PA, Wells A : Gene expression patterns in isolated keloid fibroblasts. Wound Repair Regen 2006; 14: 463–470.
Seifert O, Bayat A, Geffers R et al: Identification of unique gene expression patterns within different lesional sites of keloids. Wound Repair Regen 2008; 16: 254–265.
Smith JC, Boone BE, Opalenik SR, Williams SM, Russell SB : Gene profiling of keloid fibroblasts shows altered expression in multiple fibrosis-associated pathways. J Invest Dermatol 2008; 128: 1298–1310.
Okino N, He X, Gatt S, Sandhoff K, Ito M, Schuchman EH : The reverse activity of human acid ceramidase. J Biol Chem 2003; 278: 29948–29953.
Coant N, Sakamoto W, Mao C, Hannun YA : Ceramidases, roles in sphingolipid metabolism and in health and disease. Adv Biol Regul 2016; 63: 122–131.
O'Connell JR, Weeks DE : PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 1998; 63: 259–266.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR : Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002; 30: 97–101.
Fishelson M, Geiger D : Exact genetic linkage computations for general pedigrees. Bioinformatics 2002; 18 (Suppl 1): S189–S198.
Sobel E, Lange K : Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet 1996; 58: 1323–1337.
Santos-Cortez RL, Faridi R, Rehman AU et al: Autosomal-recessive hearing impairment due to rare missense variants within S1PR2. Am J Hum Genet 2016; 98: 331–338.
Li H, Durbin R : Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760.
McKenna A, Hanna M, Banks E et al: The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–1303.
Wang K, Li M, Hakonarson H : ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38: e164.
Liu X, Wu C, Li C, Boerwinkle E : dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat 2016; 37: 235–241.
Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J : A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014; 46: 310–315.
Shihab HA, Gough J, Cooper DN et al: Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat 2013; 34: 57–65.
Chun S, Fay JC : Identification of deleterious mutations within three human genomes. Genome Res 2009; 19: 1553–1561.
Reva B, Antipin Y, Sander C : Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res 2011; 39: e118.
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D : MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010; 7: 575–576.
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP : Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012; 7: e46688.
Ng PC, Henikoff S : Predicting deleterious amino acid substitutions. Genome Res 2001; 11: 863–874.
Adzhubei IA, Schmidt S, Peshkin L et al: A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Li CM, Park JH, He X et al: The human acid ceramidase gene (ASAH): structure, chromosomal location, mutation analysis, and expression. Genomics 1999; 62: 223–231.
Braun F, Rinschen MM, Bartels V et al: Altered lipid metabolism in the aging kidney identified by three layered omic analysis. Aging (Albany NY) 2016; 8: 441–457.
Sugita M, Dulaney JT, Moser HW : Ceramidase deficiency in Farber's disease (lipogranulomatosis). Science 1972; 178: 1100–1102.
Koch J, Gartner S, Li CM et al: Molecular cloning and characterization of a full-length complementary DNA encoding human acid ceramidase. Identification of the first molecular lesion causing Farber disease. J Biol Chem 1996; 271: 33110–33115.
Bashyam MD, Chaudhary AK, Kiran M et al: Molecular analyses of novel ASAH1 mutations causing Farber lipogranulomatosis: analyses of exonic splicing enhancer inactivating mutation. Clin Genet 2014; 86: 530–538.
Alves MQ, Le Trionnaire E, Ribeiro I et al: Molecular basis of acid ceramidase deficiency in a neonatal form of Farber disease: identification of the first large deletion in ASAH1 gene. Mol Genet Metab 2013; 109: 276–281.
Kim SY, Choi SA, Lee S et al: Atypical presentation of infantile-onset farber disease with novel ASAH1 mutations. Am J Med Genet A 2016; 170: 3023–3027.
Zhou J, Tawk M, Tiziano FD et al: Spinal muscular atrophy associated with progressive myoclonic epilepsy is caused by mutations in ASAH1. Am J Hum Genet 2012; 91: 5–14.
Li CM, Park JH, Simonaro CM et al: Insertional mutagenesis of the mouse acid ceramidase gene leads to early embryonic lethality in homozygotes and progressive lipid storage disease in heterozygotes. Genomics 2002; 79: 218–224.
Nakashima M, Chung S, Takahashi A et al: A genome-wide association study identifies four susceptibility loci for keloid in the Japanese population. Nat Genet 2010; 42: 768–771.
Zhu F, Wu B, Li P et al: Association study confirmed susceptibility Loci with keloid in the Chinese Han population. PLoS ONE 2013; 8: e62377.
Marneros AG, Norris JE, Watanabe S, Reichenberger E, Olsen BR : Genome scans provide evidence for keloid susceptibility loci on chromosomes 2q23 and 7p11. J Invest Dermatol 2004; 122: 1126–1132.
Roelfsema JH, Peters DJ : Rubinstein-Taybi syndrome: clinical and molecular overview. Expert Rev Mol Med 2007; 9: 1–16.
Campaner AB, Ferreira LM, Gragnani A, Bruder JM, Cusick JL, Morgan JR : Upregulation of TGF-beta1 expression may be necessary but is not sufficient for excessive scarring. J Invest Dermatol 2006; 126: 1168–1176.
Louw L, Engelbrecht AM, Cloete F, van der Westhuizen JP, Dumas L : Impairment in the fatty acid composition of keloids. Adv Exp Med Biol 1997; 400B: 905–910.
Tachi M, Iwamori M : Mass spectrometric characterization of cholesterol esters and wax esters in epidermis of fetal, adult and keloidal human skin. Exp Dermatol 2008; 17: 318–323.
Lu F, Gao J, Ogawa R, Hyakusoku H, Ou C : Fas-mediated apoptotic signal transduction in keloid and hypertrophic scar. Plast Reconstr Surg 2007; 119: 1714–1721.
Ishihara H, Yoshimoto H, Fujioka M et al: Keloid fibroblasts resist ceramide-induced apoptosis by overexpression of insulin-like growth factor I receptor. J Invest Dermatol 2000; 115: 1065–1071.
Burck U, Moser HW, Goebel HH, Gruttner R, Held KR : A case of lipogranulomatosis Farber: some clinical and ultrastructural aspects. Eur J Pediatr 1985; 143: 203–208.
Zappatini-Tommasi L, Dumontel C, Guibaud P, Girod C : Farber disease: an ultrastructural study. Report of a case and review of the literature. Virchows Arch A Pathol Anat Histopathol 1992; 420: 281–290.
Dulaney JT, Milunsky A, Sidbury JB, Hobolth N, Moser HW : Diagnosis of lipogranulomatosis (Farber disease) by use of cultured fibroblasts. J Pediatr 1976; 89: 59–61.
Schuchman EH : Acid ceramidase and the treatment of ceramide diseases: the expanding role of enzyme replacement therapy. Biochim Biophys Acta 2016; 1862: 1459–1471.
Acknowledgements
We are indebted to the family members who participated in this study. We thank the recruitment teams in Nigeria (A Fatokun, E Josiah, E Atiba, P Adebayo, K Adefila, P Ilesanmi and M Olaleye) for their efforts. This study was supported by the National Institutes of Health (NIH) through the National Center for Research Resources grant M01RR006192 to the Clinical Research Center at UCHC, and National Institute of Arthritis and Musculoskeletal and Skin Diseases grant R01AR45286 to ER. Exome sequencing at the UWCMG was funded by NIH – National Human Genome Research Institute and National Heart, Lung and Blood Institute grant UM1-HG006493 to DAN, MJB and SML.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Santos-Cortez, R., Hu, Y., Sun, F. et al. Identification of ASAH1 as a susceptibility gene for familial keloids. Eur J Hum Genet 25, 1155–1161 (2017). https://doi.org/10.1038/ejhg.2017.121
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.121
This article is cited by
-
Mesenchymal stem cell therapy in hypertrophic and keloid scars
Cell and Tissue Research (2021)
-
Acid ceramidase deficiency: Farber disease and SMA-PME
Orphanet Journal of Rare Diseases (2018)
