
Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is the most common monogenic kidney disorder and is due to disease-causing variants in PKD1 or PKD2. Strong genotype–phenotype correlation exists although diagnostic sequencing is not part of routine clinical practice. This is because PKD1 bears 97.7% sequence similarity with six pseudogenes, requiring laborious and error-prone long-range PCR and Sanger sequencing to overcome. We hypothesised that whole-genome sequencing (WGS) would be able to overcome the problem of this sequence homology, because of 150 bp, paired-end reads and avoidance of capture bias that arises from targeted sequencing. We prospectively recruited a cohort of 28 unique pedigrees with ADPKD phenotype. Standard DNA extraction, library preparation and WGS were performed using Illumina HiSeq X and variants were classified following standard guidelines. Molecular diagnosis was made in 24 patients (86%), with 100% variant confirmation by current gold standard of long-range PCR and Sanger sequencing. We demonstrated unique alignment of sequencing reads over the pseudogene-homologous region. In addition to identifying function-affecting single-nucleotide variants and indels, we identified single- and multi-exon deletions affecting PKD1 and PKD2, which would have been challenging to identify using exome sequencing. We report the first use of WGS to diagnose ADPKD. This method overcomes pseudogene homology, provides uniform coverage, detects all variant types in a single test and is less labour-intensive than current techniques. This technique is translatable to a diagnostic setting, allows clinicians to make better-informed management decisions and has implications for other disease groups that are challenged by regions of confounding sequence homology.
Similar content being viewed by others

Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is a common monogenic disorder with a prevalence of at least 1 in 1000.1 The disorder results in the formation of renal cysts and subsequently leads to end-stage kidney disease (ESKD) that requires dialysis or transplantation.2 ADPKD is caused by disease-causing variants in either the PKD1 or PKD2 genes, with 85% of patients having disease-causing variants in PKD1.1 In general, patients with disease-causing variants in PKD1 develop ESKD 20 years earlier than those with disease-causing variants in PKD2.1 In clinical practice, a diagnosis of ADPKD is usually made in adulthood, after significant disease progression, using imaging-based phenotypic criteria. Despite the prognostic information that can be gained from knowledge of genotype, it is not current routine clinical practice to obtain a molecular diagnosis in patients with ADPKD.
PKD1 is a 47.2 Kb gene comprising 46 exons. Exons 1–33 share, on average, 97.7% sequence similarity to six pseudogenes that lie proximal to PKD1 on chromosome 16.3, 4 These pseudogenes have arisen through successive segmental genome duplication events during recent primate evolution.3, 4 The presence of pseudogenes has made it difficult to develop cost-effective, accurate sequencing methods for PKD1. To date, long-range PCR (LR-PCR) amplification, followed by Sanger sequencing, has been the gold standard used by most diagnostic laboratories.1, 5, 6 This technique has a diagnostic rate of approximately 90% in well-phenotyped trial cohorts and 40–60% in the commercial reference laboratory.7, 8, 9 The technique is labour intensive and thus expensive. More recently, targeted massively parallel sequencing (MPS) has been used to sequence PKD1 and PKD2.5, 10, 11, 12 An amplicon-based strategy using LR-PCR amplification of PKD1 and PKD2 exons, followed by MPS obtained a diagnostic rate of ~60% in cohorts selected using only imaging criteria (ie, for whom a disease-causing variant was not already known).5, 10, 11, 12 These techniques are laborious and the PCR amplification process is error prone. Capture-based strategies have been trialled, to enrich for PKD1 and PKD2 exons, and in a small discovery cohort (n=12) the diagnostic rate was 83%.11 Capture-based approaches are biased against capturing exons with high GC content, and even when a very high average sequencing depth per base is achieved, there is still a significant fraction of bases with insufficient coverage to make a variant call.13 Furthermore, it is difficult to design short (65 nt) oligonucleotide capture probes that avoid selecting PKD1 pseudogenes.11, 12 Finally, the use of custom-designed capture probes requires pooling of samples to be cost effective, which may add significant delays in the clinical setting. PKD2 is a 68.0 kb gene in chromosome 4, comprising 15 exons. PKD2 does not have any associated pseudogenes and is relatively more straightforward to sequence.
Whole-genome sequencing (WGS) uses straightforward DNA extraction and library preparation and sequences the entire genome without distinction to coding or noncoding status.14 Sequencing the entire genome avoids the capture bias associated with targeted sequencing, and typically has a far more uniform genome-wide depth of coverage. This affords broad power to detect single-nucleotide variants (SNVs) and small insertions and deletions (indels), and the ability to detect larger copy number variants (CNVs) and structural variants (SVs), such as inversions and translocations.15, 16 Since the introduction of the Illumina HiSeq X sequencing system, the cost of WGS has decreased significantly and is likely to continue to reduce in cost over time.
We hypothesised that WGS, with its avoidance of capture bias, uniform coverage and longer read length would be better able to detect disease-causing variants in PKD1 and PKD2, particularly in the pseudogene-homologous region. In this prospective study, we performed WGS on 28 patients with ADPKD, and identified disease-causing variants in 86% of these patients. This is the first application of WGS to an ADPKD cohort and we demonstrate that this technique is a reliable and reproducible method with which to overcome sequence homology and obtain a molecular diagnosis in ADPKD patients.
Materials and methods
Patients, over the age of 18, with a diagnosis of ADPKD made based on standard clinical and imaging criteria17 were prospectively recruited into the study. For those without a family history of ADPKD, a presumptive diagnosis of ADPKD was made if multiple bilateral renal cysts were seen on ultrasound images and there were no manifestations suggestive of another cystic kidney disease.18 None of the patients had undergone diagnostic sequencing previously and all patients were from unique pedigrees. A cohort based on phenotype, rather than a previous molecular diagnosis using an alternate diagnostic technique, was selected, given this would avoid any potential technical bias and offer a more ‘real world’ cohort. Ethics approval for the study was obtained from the St Vincent’s Hospital Human Research Ethics Committee (HREC/13/SVH/119). All participants provided written consent.
Genomic DNA was extracted from peripheral blood lymphocytes using standard protocols, sheared to 350 bp, and a sequencing library for each patient was created using the TruSeq Nano DNA HT Sample Prep Kit (Illumina Inc., California, CA, USA). Following clustering of each library on a single lane of a V1 patterned flowcell, paired-end sequencing with 150 bp read length was performed using the Illumina HiSeq X, within the Kinghorn Centre for Clinical Genomics, at the Garvan Institute of Medical Research, Sydney, Australia.
Raw fastq files were transferred to DNAnexus (www.dnanexus.com), a cloud-based genomic analysis platform, utilising Amazon Web Services. Paired-end short reads were aligned to the hs37d5 reference genome using BWA MEM (v0.7.10-r789) and sorted and PCR duplicates marked with novosort (v1.03.01).19 In one sample, BAM files from two lanes were merged using novosort. The reference sequence used was the 1000 Genomes Phase 2 reference genome, which comprises the GRCh37 reference genome, including decoy sequences and the human herpesvirus 4 type 1 (hs37d5).
A Base Quality (BQ) score was generated for each base sequenced and was used to give a measure of the probability that the base called at that point is the true base. A Mapping Quality (MQ) score was assigned to each read. Reads that align to multiple parts of the genome are given a MQ score of 0, and are filtered out from calculation of depth of coverage and variant calling (see below). Each variant called was given a QUAL score, that is, a measure of the likelihood that the variant is present in the cohort. Finally, a Genotype Quality (GQ) score is obtained, which is a measure of the probability that the genotype of the particular patient at that allele is correct. QUAL and GQ scores incorporate the BQ and MQ scores for each base and read that comprises that variant as well as the depth of coverage.
Following the GATK best practices guide, reads were realigned around indels, and BQ scores recalibrated to improve the quality of the alignments, using GATK (v3.3).20 SNV and short (<50 bp) indels were identified using a GATK HaplotypeCaller, in GVCF mode. Data were subset to coding exons +10 bp as defined by CCDS v19, to reduce computation time. Variants from all samples were then joint variant called using GATK GenotypeGVCFs. Variant Quality Score Recalibration was applied to annotate variants as passing all filters. VCF files were then imported into GenePool (www.stationxinc.com) for variant annotation, filtration and interpretation. Variants passing all filters, with depth>10 and GQ>30, were considered. Genome-wide coverage was assessed using the Illumina ‘Sequencing Coverage Calculation Methods for Human WGS’ Technical Note.21 Per-exon coverage was calculated using GATK DepthOfCoverage, based on CCDS exons +10 bp on either side to account for splice regions, with MQ>20 and BQ>20.
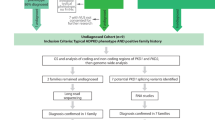
Variant filtering and interpretation were performed in a targeted manner towards PKD1 and PKD2, according to the flow-chart described in Figure 1. The pathway was modified from the American College of Medical Genetics (ACMG) guidelines for interpretation of sequence variants.22 All variants were classified as affects function, likely affects function, does not affect function, likely does not affect function or of uncertain significance. In addition to this, variants were classified as hypomorphic if classified as such in the Polycystic Kidney Disease Mutation Database (PKDB).23 In silico analysis tools that assessed conservation and splicing impact were used to assess all synonymous and missense variants with an allele frequency <5% (Mutation Taster, Polyphen2).

Variant pathogenicity classification algorithm. Algorithm used to classify pathogenicity of every variant identified. Modified from guidelines issued by American College of Medical Genetics and Genomics.
Data from patients who did not have a disease-causing SNP identified in PKD1 and PKD2 were re-analysed in order to assess for rearrangements or CNVs. SV were identified from split reads and discordant pairs using lumpy v0.2.1124 and CNV from read depth differences using CNVnator v0.325 as implemented in the SpeedSeq pipeline 0.0.3a.26
In patients in whom a disease-causing variant in PKD1 or PKD2 could not be identified, seven additional genes (HNF1B, PKHD1, SEC63, PRKCSH, TSC1, TSC2, OFD1) that are also associated with a polycystic kidney phenotype were assessed for SNV and indel, and HNF1B was assessed for deletions. In addition, in undiagnosed patients, the criteria for identifying SNVs and small indels was relaxed to include reads with MQ and BQ >10.
Sanger sequencing was performed to confirm the existence of all function-affecting and likely function-affecting variants identified via WGS. For variants within the PKD1 homologous region (exons 1–33), the pertinent regions were first amplified using LR-PCR. LR-PCR primers were generated and amplification was performed using the method previously described by Tan et al.10 In patients in whom a disease-causing variant was not identified, any exons with a depth of coverage <10 were Sanger sequenced. MLPA was performed to confirm the existence of exonic deletions. The commercial ‘MRC-Holland SALSA MLPA probemix P351-C1/P352-D1 PKD1-PKD2′ MLPA kit was used according to the method outlined by the manufacturer. All variants were submitted to the publically available PKDB (http://pkd.mayo.edu).
Results
We prospectively recruited a cohort of 28 unrelated patients who met the established diagnostic criteria for ADPKD. The cohort consisted of 14 males and 14 females whose ages ranged between 31 and 85, where ESKD had been reached in 16 patients. We performed WGS using the Illumina HiSeq X on all 28 samples, generating a total of 3680 Gb of sequencing output. On average, we obtained 131.4 Gb (range 126.3–134.0) per patient. On average, 86.9% (range 77.2–93.5%) of bases had BQ greater than 30 (ie, 0.001 probability of an error; Supplementary Table S1). The average genome-wide depth of coverage per patient was 31.9 reads (range 28.4–36.1), with 22/28 samples having a mean depth of coverage of greater than 30 (Supplementary Table S1).
Average sequencing coverage for the coding exons of PKD1 (29.8, 95% CI 28.7–30.9) was similar to PKD2 (31.1, 95% CI 30.1–32.1), however with slightly higher variability, likely due to the pseudogenes (Table 1). Despite the lower average sequencing depth of WGS relative to targeted sequencing, the uniform depth of coverage from WGS and no capture bias typically results in higher proportions of targeted bases with sufficient depth to detect variants. Accordingly, the percentage of all coding bases in PKD1 or PKD2 covered to at least 15 times read depth was high, at 97.3% (95% CI 96.7–97.9) and 98.5% (95% CI 97.9–99.1) for PKD1 and PKD2, respectively, suggesting >99% power to detect heterozygous variants within the majority of each gene13 (Table 1). Similarly high depth and breadth of coverage were observed in PKD1 and PKD2 introns (data not shown).
We also assessed the proportion of bases sequenced to greater than 15 times read depth in each exon (Figure 2a). In all, 92.0% of exons had 100% of their bases sequenced to a depth of at least 15 times, and 94.8% had at least 95% of their bases sequenced at least 15 times. Exons 1, 42 and 43 of PKD1 and exon 1 of PKD2 had consistently lower coverage. These are the only four exons that have >70% GC content (red line, Figure 2a). There was no noticeable difference in coverage over the exons that are homologous with PKD1 pseudogenes (ie exons 1–33) relative to those that are unique (exons 34–46).

Coverage and Mapping Quality (MQ) for PKD1, PKD2 and pseudogenes. (a) Coverage across PKD1 and PKD2. The percentage of bases covered with at least 15 unique reads for each exon for PKD1 and PKD2 is shown ±95% confidence interval. The bars are coloured proportionally to the percentage covered. The average GC content of each exon is also shown (red line) on the same scale. (b) MQ for PKD1, PKD2 and pseudogenes. MQ is a measure of the likelihood that a particular read is aligned to the correct segment of the genome. For BWA MEM, the sequence aligner that we used, the maximum possible MQ is 60. The average mapping quality ±95% confidence interval, from n=6 patients are shown (patient ids: 506, 547, 609, 610, 625, 626).
Given the concerns about pseudogene homology potentially confounding the sequencing, we assessed the average MQ of the read alignment over PKD1, PKD2, and the six PKD1 pseudogenes (Figure 2b). MQ is a measure of the likelihood that a particular read is aligned to the correct segment of the genome. The average MQ for PKD2 was 60.0, which is the maximum possible value. The average MQ for PKD1 was 54.4, which is substantially higher than the 4.4–35.7 observed for the other pseudogenes, that is, the reads were 74–10 000 × more confidently aligned to PKD1 than to each of the pseudogenes (Figure 2b). Supplementary Figure S1 shows the high frequency of reads with MQ=0, in pseudogene PKD1P4, relative to PKD1.
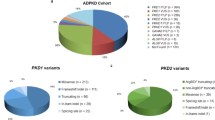
Across the cohort, we identified 183 SNVs and small insertions or deletions within PKD1 and PKD2 exons and flanking splice regions (Supplementary Table S2). An average of 6.5 variants was identified per patient (range 2–22). Most variants (85%) were identified in PKD1. The variants consisted of 88 (48%) synonymous, 72 (40%) missense, 9 (5%) nonsense, 8 (4%) frameshift and 4 (2%) splice-site variants (Figure 3, left). In addition to these SNVs and small insertions or deletions, we identified two (1%) large heterozygous deletions. A 5461 bp deletion was detected in PKD2 (chr4: 88,952,071–88,957,532), deleting exon 3, which had support from split reads, discordant pairs and reduced read depth (Figure 4). A 2199 bp deletion was detected in PKD1 (chr16: 2,146,901–2,149,100), deleting exons 31–34, which had support from reduced read depth and discordant pairs.

Types of variants detected in PKD1 and PKD2 via whole-genome sequencing. The pathogenicity rating is based on current ACMG guidelines, described in more detail in Figure 1. VOUS, variant of uncertain significance.

Single-exon PKD2 deletion detected with whole-genome sequencing. A 5461 bp base deletion, which overlaps exon 3 of PKD2. Three lines of evidence support the heterozygous deletion: the reduction in read depth to ~50% of the surrounding regions; the presence of three spanning read pairs where each read aligned to either side of the deletion; and the presence of four split reads, where the start and end of the sequencing reads are on either side of the breakpoint.
All variants were classified in accordance with the ACMG guidelines for interpretation of sequence variants and variants of all pathogenicity types were identified (Figure 3, right). We obtained a molecular diagnosis of ADPKD in 24 of 28 (86%) patients (Table 2). In total, 10 deletions were identified, ranging from deletion of a single nucleotide to 5461 nucleotides. Both large deletions disrupt the reading frame of the resulting protein, and both had not been reported in the database of genomic variation,27 thus we classified them as loss of function. Most, 17 (71%) of the disease-causing variants were within PKD1. The majority of these variants (58%) have not been reported in the PKDB.23 Of the 17 PKD1 disease-causing variants, 12 (71%) were within the pseudogene-homologous region (exons 1–33). All function-affecting and likely-function-affecting variants were confirmed by LR-PCR and Sanger sequencing, and no false-positives were identified (Table 2). The exonic deletions identified in patient 506 and patient 626 were confirmed by MLPA.
WGS was performed in three additional patients who were related to a member of the original study cohort, in order to assess reproducibility of the technique. In all three cases, the familial variant was identified. Segregation analysis was performed using Sanger sequencing in five additional pedigrees, and confirmed segregation with phenotype.
In the four patients who were still undiagnosed, we relaxed the stringent criteria for identifying SNVs and small indels and identified no additional disease-causing variants. In addition, we performed Sanger sequencing of exon 1 of PKD1 in these patients, and were unable to detect additional disease-causing variants. In one of these four patients, in silico pathogenicity prediction analysis (see Materials and methods) predicted a novel missense variant (NM_001009944.2:c.11539A>C) (p.(Ser3847Arg)) to be likely pathogenic; however, in the absence of segregation and functional studies we classified this as a variant of uncertain significance. Finally, we examined additional genes (HNF1B, PKHD1, SEC63, PRKCSH, TSC1, TSC2, OFD1), also known to be associated with a cystic kidney disease phenotype, and no additional function-affecting or likely-function-affecting variants were found.
Discussion
A molecular diagnosis is not routinely made in patients with ADPKD or at risk family members, as it is difficult to sequence PKD1 accurately using currently available techniques, given the technical challenges created by the existence of six pseudogenes proximal to PKD1.5, 6 In this study we demonstrate that WGS is a reliable, minimally labour-intensive and reproducible technique with which to overcome the challenge of pseudogene homology and thus make a molecular diagnosis of ADPKD. Using this technique we were able to make a molecular diagnosis in 86% of patients.
WGS has the advantage of avoiding any capture biases due to the potentially error-prone enrichment process that is required in whole-exome sequencing and targeted MPS. In addition, because of the more uniform coverage obtained, WGS can achieve similar sensitivity with a lower average depth of coverage than is required with exome sequencing.13, 28 WGS can achieve 99% sensitivity to detect heterozygous SNVs with 22 × average depth, compared with exome sequencing which requires at least 88 × average depth to achieve similar sensitivity.13 Furthermore, the broad, uniform depth of sequencing coverage from WGS also makes the identification of copy number and structural variants far more straightforward than targeted approaches where the depth of coverage varies substantially along a given gene. Our results demonstrate that from a single lane of sequencing on modern Illumina HiSeq X instruments, we can achieve a mean depth of coverage of approximately 30 reads across the entire genome, and for PKD1, PKD2 in each patient. This uniform depth of coverage was also consistent along the pseudogene-homologous region of PKD1.
Coverage was reduced across exons 1, 42 and 43 of PKD1 and exon 1 of PKD2 and inversely correlated with GC content, which is consistent with, albeit less extreme, than previous studies using capture-based MPS.11, 12 As three of these exons fall outside of the pseudogene region, we attribute this lower sequencing depth to GC content rather than homology to the pseudogenes. Reduced coverage due to high GC content is likely due to PCR amplification bias during library creation and clonal amplification on the flowcell surface.29 Given the variability in coverage over these regions, it is possible that variants within these exons are missed with this current technique. PCR-free WGS methods are now available, and their use will avoid upfront PCR bias during library preparation and should improve coverage over these challenging exons. Despite the reduced coverage, 31 variants (17%) were detected across these regions. In addition, in the four patients in whom a disease-causing variant was not identified, aside from exon 1 of PKD1 (which was subsequently Sanger sequenced), these patients had 100% coverage to a depth of at least 10 in all exons, except in one patient where there was 94% coverage to a depth of at least 10 in PKD1 exon 42, thus making the probability of a false negative result low.
This study demonstrates that WGS is not confounded by homology between the pseudogene region and PKD1. Manual review of the mapping quality over PKD1 revealed that the majority of the reads that aligned to the PKD1 region were mapping uniquely, whereas the majority of reads in the PKD1 pseudogenes had poor mapping quality. This suggests that the latest WGS technology, with 150 bp paired-end reads, can discriminate PKD1 as being genetically distinct from the pseudogenes, but that the pseudogenes are more closely related, and thus hard to distinguish from each other, consistent with previous evolutionary analyses.4, 30 This is likely due to gene conversion events that occur between the more closely located pseudogenes. The uniformly high mapping quality of short reads within PKD1 suggests that variants we identified have a high likelihood of being real. We hypothesise that more uniform coverage, along with the 150 bp paired-end read length and the ability to sequence the intronic regions, has resulted in more accurate alignment of reads between the pseudogene regions and PKD1.
In this cohort, we identified a much higher proportion of nonsense mutations, and in particular, essential splice-site mutations than reported either in PKDB or in previous reports using targeted sequencing. Of 24 variants, 23 (96%) had a nonsense variant or large deletion, whereas PKDB contains 23% missense variants annotated as function-affecting, with a note from the curators of the database that the pathogenicity of most of the missense variants has not been proven.23 We attribute the higher proportion of nonsense mutations in our cohort to our careful clinical inclusion criteria, the broad coverage of WGS which comprehensively sequences the entire gene, and that many of the previously reported missense variants may simply be the most damaging variant that was found in a ADPKD patient, using a less sensitive technique. By WGS, we identified essential splice-site variants in 4/24 (16.7%) diagnoses, whereas in previous clinical cohorts that underwent targeted exome sequencing, no disease-causing splice-site variants were detected within one group, and 6 from 230 (2.61%) in another.5, 10 The numbers are small, but this suggests that WGS can obtain a high diagnostic yield in intronic regions that have lower coverage by capture-based approaches. We did not identify any single-nucleotide insertions within our cohort. The PKDB reports two patients with single-nucleotide insertions within its entire large data set, and therefore we hypothesise that due to our smaller cohort size, this variant type was not present. We did identify single-nucleotide deletions.
Our technique was also able to identify structural variants, which was demonstrated by the detection of large exonic deletions in two of our patients. Multiple lines of evidence supported the deletions: reduction of read depth, reads that span the deletion, and in the case of the PKD2 deletion, split reads, where each half of the read maps to either side of the breakpoint. These breakpoints were in introns, and would have been missed by exome-capture-based MPS. This is pertinent for PKD1 and PKD2 variants, as small and large deletions and duplications comprise approximately 10% of pathogenic variants recorded in the ADPKD database, and are typically identified using a separate sequencing technique, MLPA, in diagnostic labs.23
The genetic diagnosis in a minority (4/28) of our patients remains unclear. One patient had a novel missense variant that possibly affects function, but without functional or segregation studies, there is insufficient evidence to classify this variant as likely to affect function. We did not identify disease-causing variants in the remaining three patients. A number of possibilities could explain these findings. First, it is possible that these patients do not have ADPKD. Second, there could be somatic mosaicism. Third, there could be regulatory variants affecting the expression levels of PKD1 or PKD2. Fourth is the potential that we have missed a disease-causing variant due to misclassification, or our incomplete understanding of the impact of particular variants. The benefit of WGS lies in the ability to further scrutinise these patients’ data for new disease genes or regulatory variants.
Our small cohort size is a potential limitation, though the sample size is comparable to other recent disease-cohort based WGS studies and is reflective of the prospective study design.15, 16 Our study is however the first WGS study in any nephrology cohort.
There is important clinical utility in obtaining a genetic diagnosis of ADPKD. A clinical diagnosis can be difficult to make in young patients, those without a family history and those with relatively fewer cysts on imaging studies.17 A more cost-effective and accurate molecular diagnostic test, which this technique has the potential to offer, would provide diagnostic certainty for this cohort of patients, thus avoiding serial imaging and periods of diagnostic uncertainty. A genetic diagnosis also adds clarity in the setting of living-related kidney donation, ensuring a phenotypically normal donor does not carry a pathogenic familial variant.31 Sample preparation for WGS is a streamlined process, making it cost-favourable to current testing modalities that require labour-intensive laboratory preparation and cost of WGS is expected to decrease further. The ability to detect CNV with this technique also negates the need for additional cost for CNV analysis that current diagnostic methods require.
Current prenatal diagnostic techniques require prior knowledge of the familial mutation. More accessible genetic diagnostic techniques will assist in the increased uptake of these techniques among ADPKD families.
There is clear research potential in WGS of ADPKD cohorts. The improved coverage and sequencing of intronic regions will allow analysis of modifier genes and the identification of other genes associated with the ADPKD phenotype. Improved genetic diagnostic techniques will better characterise ADPKD trial cohorts, and allow correlation of progression and response to treatment with genotype. The ability to cost effectively make a genetic diagnosis prior to the onset of a clinical phenotype allows the opportunity for treatment trials that can offer an intervention at the early stages of cyst development, rather than current trials that are directed at treating patients who have already undergone significant disease progression. The ability to overcome the difficulties of pseudogene homology also has impacts for other disease groups that also have similar sequencing challenges due to sequence homology.
We have demonstrated a strong diagnostic yield for WGS in an ADPKD cohort and demonstrate the first sequencing method that can detect all types of disease-causing variants in ADPKD using a single test. This finding requires further characterisation in a larger cohort, which could now be justified given these results, and offers the potential to improve patients’ access to a genetic diagnosis and thus better tailor prognostic information and management decisions.
References
Cornec-Le Gall E, Audrezet MP, Chen JM et al: Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol 2013; 24: 1006–1013.
Cornec-Le Gall E, Audrézet M-P, Le Meur Y, Chen J-M, Férec C : Genetics and pathogenesis of autosomal dominant polycystic kidney disease: 20 years on. Hum Mutat 2014; 35: 1393–1406.
Kirsch S, Pasantes J, Wolf A et al: Chromosomal evolution of the PKD1 gene family in primates. BMC Evol Biol 2008; 8: 263.
Symmons O, Varadi A, Aranyi T : How segmental duplications shape our genome: recent evolution of ABCC6 and PKD1 Mendelian disease genes. Mol Biol Evol 2008; 25: 2601–2613.
Rossetti S, Hopp K, Sikkink RA et al: Identification of gene mutations in autosomal dominant polycystic kidney disease through targeted resequencing. J Am Soc Nephrol 2012; 23: 915–933.
Rossetti S, Strmecki L, Gamble V, Komel R, Winearls CG : Mutation analysis of the entire PKD1 gene: genetic and diagnostic implications. Am J Hum Genet 2001; 68: 46–63.
Tan AY, Blumenfeld J, Michaeel A et al: Autosomal dominant polycystic kidney disease caused by somatic and germline mosaicism. Clin Genet 2014; 87: 373–377.
Rossetti S, Consugar MB, Chapman AB et al: Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18: 2143–2160.
Audrézet M-P, Cornec-Le Gall E, Chen J-M et al: Autosomal dominant polycystic kidney disease: comprehensive mutation analysis of PKD1 and PKD2 in 700 unrelated patients. Hum Mutat 2012; 33: 1239–1250.
Tan AY, Michaeel A, Liu G et al: Molecular diagnosis of autosomal dominant polycystic kidney disease using next-generation sequencing. J Mol Diagn 2014; 16: 216–228.
Trujillano D, Bullich G, Ossowski S et al: Diagnosis of autosomal dominant polycystic kidney disease using efficient PKD1and PKD2targeted next-generation sequencing. Mol Genet Genomic Med 2014; 2: 412–421.
Eisenberger T, Decker C, Hiersche M et al: An efficient and comprehensive strategy for genetic diagnostics of polycystic kidney disease. PLoS One 2015; 10: e0116680.
Meynert A, Ansari M, FitzPatrick D, Taylor M : Variant detection sensitivity and biases in whole genome and exome sequencing. BMC Bioinformatics 2014; 15: 247–258.
Biesecker LG, Green RC : Diagnostic clinical genome and exome sequencing. N Engl J Med. 2014; 370: 2418–2425.
Nishiguchi K, Tearle R, Liu Y, Oh E, Katsanis N, Rivolta C : Whole genome sequencing in patients with retinitis pigmentosa reveals pathogenic DNA structural changes and NEK2 as a new disease gene. Proc Natl Acad Sci USA 2013; 10: 16139–16144.
Gilissen C, Hehir-Kwa JY, Thung DT et al: Genome sequencing identifies major causes of severe intellectual disability. Nature 2014; 511: 344–347.
Pei Y, Obaji J, Dupuis A et al: Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol 2009; 20: 205–212.
Harris PC, Torres VE : Polycystic kidney disease, autosomal dominant; in Pagon RA, Adam MP, Ardinger HH (eds): Seattle: University of Washington, 2015. Available at http://www.ncbi.nlm.nih.gov/books/NBK1246/.
Li H . Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013. Available at http://arxiv.org/abs/1303.3997.
DePristo MA, Banks E, Poplin R et al: A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011; 43: 491–498.
Illumina. Sequencing coverage calculation methods for human whole-genome sequencing. 2014:1–2. Available at http://www.illumina.com/content/dam/illumina-marketing/documents/products/technotes/hiseq-x-30x-coverage-technical-note-770-2014-042.pdf.
Richards S, Aziz N, Bale S et al: Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–424.
ADPKD Mutation Database: PKDB. Available at http://pkdb.pkdcure.org (accessed 30 April 2015).
Layer RM, Chiang C, Quinlan AR, Hall IM : LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 2014; 15: 1–19.
Abyzov A, Urban AE, Snyder M, Gerstein M : CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 2011; 21: 974–984.
Chiang C, Layer RM, Faust GG et al: SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat Methods 2015; 12: 966–968.
MacDonald JR, Ziman R, Yuen RKC, Feuk L, Scherer SW : The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res 2013; 42: D986–D992.
Kingsmore S, Saunders C : Deep sequencing of patient genomes for disease diagnosis: when will it become routine? Sci Transl Med 2011; 3: 23–27.
Aird D, Ross MG, Chen W-S et al: Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol 2011; 12: R18.
Bogdanova N, Markoff A, Gerke V, McCluskey M, Horst J, Dworniczak B : Homologues to the first gene for autosomal dominant polycystic kidney disease are pseudogenes. Genomics 2001; 74: 333–341.
Simms R, Travis D, Durkie M, Wilson G, Dalton A, Ong A : Genetic testing in the assessment of living related kidney donors at risk of autosomal dominant polycystic kidney disease. Transplantation 2014; 99: 1023–1029.
Acknowledgements
Many thanks for the patients and families that participated in the study. MJC is supported by a Cancer Institute NSW early career fellowship (13/ECF/1-46). We acknowledge financial support from the Burcher, Kirkpatrick and Lewis families, and the Kinghorn Foundation, without which this research would not have been possible.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Mallawaarachchi, A., Hort, Y., Cowley, M. et al. Whole-genome sequencing overcomes pseudogene homology to diagnose autosomal dominant polycystic kidney disease. Eur J Hum Genet 24, 1584–1590 (2016). https://doi.org/10.1038/ejhg.2016.48
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.48
This article is cited by
-
Atypical splicing variants in PKD1 explain most undiagnosed typical familial ADPKD
npj Genomic Medicine (2023)
-
Identification of novel single-nucleotide variants altering RNA splicing of PKD1 and PKD2
Journal of Human Genetics (2022)
-
Recommendations for whole genome sequencing in diagnostics for rare diseases
European Journal of Human Genetics (2022)
-
Identification of ACOT13 and PTGER2 as novel candidate genes of autosomal dominant polycystic kidney disease through whole exome sequencing
European Journal of Medical Research (2021)
-
Genomic diagnostics in polycystic kidney disease: an assessment of real-world use of whole-genome sequencing
European Journal of Human Genetics (2021)
