
Abstract
Time to fall asleep (sleep latency) is a major determinant of sleep quality. Chronic, long sleep latency is a major characteristic of sleep-onset insomnia and/or delayed sleep phase syndrome. In this study we aimed to discover common polymorphisms that contribute to the genetics of sleep latency. We performed a meta-analysis of genome-wide association studies (GWAS) including 2 572 737 single nucleotide polymorphisms (SNPs) established in seven European cohorts including 4242 individuals. We found a cluster of three highly correlated variants (rs9900428, rs9907432 and rs7211029) in the RNA-binding protein fox-1 homolog 3 gene (RBFOX3) associated with sleep latency (P-values=5.77 × 10−08, 6.59 × 10−08 and 9.17 × 10−08). These SNPs were replicated in up to 12 independent populations including 30 377 individuals (P-values=1.5 × 10−02, 7.0 × 10−03 and 2.5 × 10−03; combined meta-analysis P-values=5.5 × 10−07, 5.4 × 10−07 and 1.0 × 10−07). A functional prediction of RBFOX3 based on co-expression with other genes shows that this gene is predominantly expressed in brain (P-value=1.4 × 10−316) and the central nervous system (P-value=7.5 × 10−321). The predicted function of RBFOX3 based on co-expression analysis with other genes shows that this gene is significantly involved in the release cycle of neurotransmitters including gamma-aminobutyric acid and various monoamines (P-values<2.9 × 10−11) that are crucial in triggering the onset of sleep. To conclude, in this first large-scale GWAS of sleep latency we report a novel association of variants in RBFOX3 gene. Further, a functional prediction of RBFOX3 supports the involvement of RBFOX3 with sleep latency.
Similar content being viewed by others


Introduction
Sleep occurs during defined periods within the 24 h cycle.1 Its timing and duration is considered to be regulated by two processes; (i) a circadian process (ie, being awake during the day and asleep during night) and (ii) a homeostatic process that represents the sleep pressure accumulating during wakefulness, ie, the longer one is awake, the greater sleep pressure and the greater will be the duration of sleep when permitted.2 Sleep latency, ie, the duration it takes to fall asleep, is a measure of sleep quality computed as the time interval between ‘lights out’ until the onset of sleep. Photic information from the retina is projected to the suprachiasmatic nucleus (SCN) via the retino-hypothalamic tract. In humans, the SCN is considered the pacemaker for the timing of daily sleep–wake behavior and consists of ~10 000 neurons located on both sides of the midline above the optic chiasma, ~3 cm behind the eyes.3, 4 At night, the SCN signals the release of melatonin – a hormone produced by the pineal gland that promotes sleep. Sleep latency may be assessed using self-reports, actigraphy or with polysomnography. Normal sleepers are known to provide estimates of sleep latency that correlate well with polysomnographic measures.5, 6 Previous studies show that sleep latency is largely independent of an individual’s chronotype.7
Inter-individual differences in sleep latency are caused by both genetic and non-genetic factors. The latter include gender,8, 9 age,8 consumption of stimulants,10 dietary intake,11 sedentary life12 and illnesses, such as depression.13 Persistent increased sleep latency is a major characteristic of delayed sleep phase syndrome14 and/or sleep-onset insomnia.15, 16, 17, 18 Prolonged sleep latency may shorten sleep duration and may lead to a wide range of problems including irritability, cognitive impairment, depression and loss of productivity as well as accident rates due to sleepiness. Increased sleep latency has also been associated with poor academic performance in children and adolescents.19 Although very long sleep latency can lead to sleep deprivation, very short sleep latency can reflect sleep deprivation (ie, ‘sleep debt’ due to insufficient sleep normally shortens sleep latency). Unusually short sleep latency also may indicate disorders of excessive sleepiness (eg, narcolepsy). When giving individuals the opportunity to sleep during the day, sleep latency is used as an objective measure of daytime sleepiness in the diagnosis of sleep disorders.
Heritability of sleep latency is estimated to be ~17–44%.20, 21, 22 Although much is known about the environmental factors that can prolong sleep latency, we know little about the genetic influences on sleep latency. In this study, we performed a meta-analysis of unpublished genome-wide association studies (GWAS) on sleep latency in order to elucidate genetic associations with this trait.
Materials and methods
Study populations (stage 1/discovery cohorts)
We meta-analyzed data from seven GWAS comprising of 4242 subjects with European ancestry (Supplementary Tables S1 and S2). The participating cohorts in the gene discovery phase included the Erasmus Rucphen Family (ERF), Estonian Genome Center (EGP/EGCUT), CROATIA-Korcula, the Micro-isolates in South Tyrol Study (MICROS), Cooperative health research in the Region of Augsburg (KORA), the Netherlands Study of Depression and Anxiety (NESDA) and the Orkney Complex Disease Study (ORCADES) (Supplementary Tables S1 and S2). A detailed description of the included studies is provided in the Supplementary text. All studies in the discovery cohort used the Munich Chronotype Questionnaire (MCTQ)23 to assess sleep latency. Subjects were asked to report how long they take to fall asleep on free and workdays. Free days sleep latency was used in the analyses of those cohorts, where a person’s sleep pattern is not influenced by professional duties24 (Supplementary Figure S1). The question from the MCTQ used to assess sleep latency (in minutes): ‘I need … minutes to fall asleep’. Sleep duration was calculated by subtracting sleep onset from sleep end. Mid-sleep was calculated as the midpoint between sleep onset and waking on free days.25 The quality control was centralized and the inclusion criteria were: (i) no use of an alarm clock on free days; (ii) no shift-work during the last 3 months; and (iii) no use of sleep medication (benzodiazepines and other pharmacological agents that influence sleep; see Supplementary Table S3). Informed consent was obtained from all study participants and an appropriate local committee approved the study protocols.
Study populations (stage 2/replication cohorts)
The replication stage included 12 independent cohorts (see Supplementary text; Supplementary Figure S1). The descriptive statistics are provided in Supplementary Table S1. Inclusion criteria were: (1) sleep assessment with any available tool and (2) No use of sleep medication (Supplementary Figure S1). Five replication cohorts (MrOS, RS-I, RS-II, RS-III and SOF) used the Pittsburg Sleep Quality Index to assess sleep (Supplementary Table S2), which uses only a single measure for sleep assessment and does not distinguish between sleep on free and working days.
Genotyping and imputation
Both discovery and replication cohorts were genotyped on a variety of platforms (Affymetrix 250 K, Illumina 318 K, Illumina 370 K, Illumina 610 k; Perlegen 600 K; Affymetrix 1000 K). Quality control was done in each group separately. The overall criteria were to exclude individuals with low call rate, excess heterozygosity and gender mismatch, and exclude variants that were out of Hardy–Weinberg equilibrium, had low minor allele frequency (MAF) or low call rate (Supplementary Table S2). In EGCUT1 study where the genome-wide data was not available, the two most significant single nucleotide polymorphisms (SNPs) for which a TaqMan assay was available were genotyped (Supplementary Table S4). Imputations of non-genotyped SNPs in the discovery cohorts were carried out within each study using either MACH26, 27 or IMPUTE,28, 29 and HapMap CEU v21a or v22 as reference (Supplementary Table S2). Genetic imputations in the replication cohorts were performed using MACH, IMPUTE, minimac or BimBam (Supplementary Table S2). Of the three SNPS rs9907432 was genotyped in most replication cohorts, whereas rs9900428 was imputed in all replication cohorts (Supplementary Table S4). The data are available in the GWAS Central database, under the accession number HGVST1836 (http://www.gwascentral.org/study/HGVST1836).
Methylation and expression
The Infinium Human Methylation 450 array (Illumina, San Diego, CA, USA) was used to quantify genome-wide DNA methylation for 748 samples from the Rotterdam study (RS-III) covering 485 577 CpG sites in the genome. Bisulfite conversion followed by amplification, hybridization and imaging were performed according to standard protocols. Illumina GenomeStudio software was used to estimate β score from intensity. Quality-control (QC) steps included removal of signal probes with a detection of P-value>0.01; and exclusion of probes with low intensity. Samples with a call rate<99% were removed from the dataset. SWAN package for R software was used to normalize remaining CpG sites and correct for batch effects.
RNA from the same samples was obtained (PAXgene) and hybridized to Illumina HumanHT-12 arrays. Raw probe intensity was obtained using BeadStudio. Gene expression data were quantile-normalized to the median distribution, and subsequently log2 transformed. The probe and sample means were centered to zero. Probes that had a detection P-value<0.05 in >10% of the samples were removed from the analysis. The final analysis included 21 328 probes, which were significantly expressed in blood.
Statistical analysis
Heritability analysis
Heritability analysis of sleep latency was performed in the family-based ERF cohort using Sequential Oligogenic Linkage Analysis Routines (SOLAR).30 SOLAR uses likelihood ratio tests to evaluate heritability by comparing a purely polygenic model with a sporadic model in the case of testing heritability. The ERF cohort forms one large family, which consists of >23 000 members spanning over 23 generations. Since this uniquely large family is too large to be analyzed with SOLAR, we cut it into smaller pedigrees (three to five generations) using the Pedcut software31 for the heritability analysis. A natural log transformation was applied to the trait before estimating the heritability. The heritability was estimated with the ‘polygenic screen’ option and using age and sex as covariates in the model. The estimate was then compared with the heritability estimate derived from the polygenic analysis of GenABEL that uses genome-wide genotype data instead of the pedigree to estimate heritability.32
Genome-wide association analysis
Individual GWAS was performed using linear regression (under additive model), natural log of sleep latency as the dependent variable, SNP allele dosage as predictor and age and sex as covariates. CROATIA-Korcula and ORCADES additionally used first three principal components as covariates in the association model. The association analyses were conducted in ProbABEL32, 33, 34 or SNPTEST.35 For cohorts with related individuals (ERF, MICROS, CROATIA-Korcula, ORCADES), a linear mixed model in ProbABEL using the ‘mmscore’ option was used to account for familial relationships. The mmscore option performs the score test that uses the inverse variance-covariance object estimated from the genetic data and returned from the ‘polygenic’ function of GenABEL32 to correct for familial relationships. This is a slightly modified FASTA method developed by Abecasis et al.36
As all three SNPs show no heterogeneity across the cohorts (P-values>0.7) (Table 1), a fixed effects meta-analysis was conducted using the inverse variance weighted method as implemented in METAL (http://www.sph.umich.edu/csg/abecasis/metal/). All SNPs that had a MAF<0.01 and low imputation quality (Rsq/proper_info<0.3) were dropped from the meta-analysis. Genomic control correction was also applied to all cohorts prior to the meta-analysis.
For the SNPs that approached genome-wide significance (P-value<5 × 10−08) we performed replication in up to 12 independent cohorts using the same model as in the initial GWAS. In the replication cohorts, among the family-based studies, ERF_ext used SOLAR, FHS used LMEKIN package of R software (http://www.r-project.org/), CROATIA-Split used the ‘mmscore’ option in ProbABEL, whereas QIMR used MERLIN to account for family structure. Other population-based cohorts used SPSS, PLINK (http://pngu.mgh.harvard.edu/purcell/plink/)37 or R software to perform association analysis (Supplementary Table S2). Meta-analysis was performed using square-root of the sample size as weights.38
Methylome-wide association analysis
Top SNPs from the meta-analysis of GWAS were tested for association with genome-wide methylation (CpG) sites by performing linear regression analyses of methylation sites on each of the three SNPs while adjusting for age, sex, technical covariates including batch effects and blood cell counts (granulocytes, lymphocytes, monocytes, erythrocytes and platelets) in R software.
Gene functional prediction and network analysis
Prediction of gene function can be conducted using a guilt-by-association approach: eg, if there are 100 genes that are known to be involved in apoptosis, identification of a gene that is strongly co-expressed with these 100 genes suggests that this gene is likely to be involved in apoptosis as well. As such co-expression data can be used to predict likely functions for genes. However, important to realize is that some phenomena exert very strong transcriptomic effects and therefore will overshadow more subtle effects. In order to be able to identify such subtle relationships as well, we conducted a principal component analysis (PCA) on an unprecedented scale.39 We collected gene expression data for three different species (homo sapiens, mus musculus and rattus norvegicus) from the Gene Expression Omnibus. We confined analyses to four different Affymetrix expression platforms (Affymetrix Human Genome U133A Array, Affymetrix Human Genome U133 Plus 2.0 Array, Affymetrix Mouse Genome 430 2.0 Array and Affymetrix Rat Genome 230 2.0 Array). For each of these platforms we downloaded the raw CEL files (20 108, 43 278, 18 639 and 6124 arrays, respectively), and used RMA for normalization. We could run RMA on all samples at once for the 20 108 Human Genome U133A Array, 18 639 Mouse Genome 430 2.0 Array and 6123 Rat Genome 230 2.0 Array. For the 43 278 Human Genome U133 Plus 2.0 Array samples we ran RMA in eight batches due to its size, by randomly assigning the samples to one of these batches. We subsequently conducted QC on the data. We first removed duplicate samples, and then conducted a PCA on the sample correlation matrix. The first principal component (PCqc) on such a matrix describes nearly always a constant pattern (dominating the data), which explains ~80–90% of the total variance.40, 41 This pattern can be regarded as probe-specific variance, independent of the biological sample hybridized to the array. The correlation of each individual microarray with this PCqc can be used to detect outliers, as arrays of lesser quality will have a lower correlation with the PCqc. We removed samples that had a correlation R<0.75. After QC in total 77 840 different samples remained for downstream analysis (54 736 human samples, 17 081 mouse samples, 6 023 rat samples). Although this QCed data set can be well used for the aforementioned guilt-by-association co-expression analysis, we reasoned that the presence of profound effects on many genes will make it difficult to identify the more subtle relationships that exist between genes. Therefore, we conducted a PCA on the probe correlation matrix, resulting in the identification of in total 2206 robustly estimated principal components (377 for Human Genome U133A, 777 for Human Genome U133 Plus 2.0, 677 for Mouse Genome 430 2.0 and 375 for Rat Genome 230 2.0) by requiring a Cronbach's alpha >0.70 for each individual principal component. Jointly these components explain between 79% and 90% of the variance in the data per Affymetrix expression platform, and many of these are well conserved across the three species.
Subsequent Gene Set Enrichment Analysis revealed that each of these 2206 components are significantly enriched (false discovery rate<0.05) for at least one GO term, KEGG, BioCarta or Reactome pathway, indicating that these components are describing biologically relevant but often diverse phenomena. While per species the very first components describe profound effects on expression (ie, many enriched pathways and GO terms), the other components are potentially equally biologically relevant, as each of the components describe certain biological phenomena. We therefore used the individual components and integrated the different platforms and species by collapsing the probe identifiers to human Ensembl genes and used orthology information from Ensembl for the mouse and rat platform, resulting in a harmonized matrix of 19 997 unique Ensembl genes × 2206 principal components.
We subsequently predicted the most likely Gene Ontology (GO) biological process using the following strategy: (i) we first ascertained each individual GO term and assessed per PC whether the genes that were explicitly annotated with this GO term showed a significant difference from the genes that were not annotated with this GO term using a t-test; (ii) we converted the resulting P-value into an 'enrichment' Z-score (to ensure normality); and (iii) we subsequently investigated RNA-binding protein fox-1 homolog 3 gene (RBFOX3) and correlated the 2206 PC eigenvector coefficients of RBFOX3 with each GO term by taking the 2206 'enrichment' Z-scores as the expression profile for that GO term. A significant positive correlation means RBFOX3 has an expression profile that is comparable to the GO term. We have visualized this method at www.genenetwork.nl/genenetwork (click on ‘Method’). In order to correct for multiple testing, we permuted Ensembl gene identifiers: using permuted data we redid the ‘enrichment’ Z-score calculation and investigated how strong RBFOX3 correlated with permuted pathway. We repeated this analysis 100 times, allowing us to determine which of these predictions were significant (controlling to the false discovery rate of 0.05). We used the same procedure to predict in which BioCarta and Reactome pathways RBFOX3 is involved.
Results
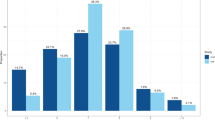
A basic description of the study populations is given in Supplementary Table S1. The heritability of sleep latency using the pedigree data of the ERF study was estimated to be 0.18 (SE=0.112, P-value=0.05) (see methods section), which is consistent with earlier findings of heritability for this trait 21 and also comparable to the heritability estimate (h2=0.20) derived from the genome-wide genotype data (see Materials and methods section for details) of the same cohort. The quantile–quantile plot of the meta-analysis shows no inflation of the chi-square statistic as the genomic control inflation factor (λ) is 1.01 (Supplementary Figure S2). The meta-analysis of GWAS produced a cluster of three borderline genome-wide significant SNPs (Table 1; Figure 1 and Supplementary Figure S3) on chromosome 17q25. The highest-ranking SNP rs9900428 hg18.chr17:g.74651323G>A) (P-value=5.7 × 10−08) had a MAF of 0.20–0.33, with the imputation quality ranging from 0.85 to 0.99 across all cohorts. The effect size (β=−0.094) and the direction of the risk were consistent across all cohorts (Table 1). The other two SNPs rs9907432 (hg18.chr17:g.74651967:G>A) (P-value=6.5 × 10−08) and rs7211029 (hg18.chr17:g.74652903C>T) (P-value=9.1 × 10−08) were in linkage disequilibrium (r2=0.95) with rs9900428 (Figure 1,Supplementary Figure S4). Figure 2 shows the mean sleep latency per genotype category for all the discovery cohorts for the three SNPs unadjusted for age and gender. Compared with the homozygote carriers of the reference allele (pooled average sleep latency=16.4 min), the homozygote carriers (pooled average sleep latency=13.5 min) of the minor allele are estimated to have a sleep latency of on average approximately 3 min less (a reduction of 18%) and the heterozygote carriers (pooled average sleep latency=15.5 min) ~1 min less (a reduction of ~6%) (Figure 2).

Regional association plot of sleep latency for the region 17q25. The plot was constructed using Locus Zoom (http://csg.sph.umich.edu/locuszoom/). The most significant SNP is depicted as a diamond and other SNPs in the region are depicted by circles. Various colors represent the extent of linkage disequilibrium with most significant SNP. The X axis gives the position in mega bases and the Y axis shows the negative logarithm of the P-values from the meta-analysis.

Means per genotype category for all the discovery cohorts for the three most significant SNPs. Black bars represent the mean sleep latency for the homozygous carriers of the effect allele. Y axis depicts the sleep latency time in minutes. Panels a, b and c represent the effects of the SNPs in individual cohorts. Panel d represents the pooled average sleep latency per genotype category across all cohorts for the three SNPs (X axis). Data used to generate this figure were not adjusted for age and sex.
We attempted to replicate these three SNPs in up to 12 independent cohorts (Supplementary Tables S2 and S4). SNP rs9900428 was available in silico in 11 cohorts, whereas rs9907432 and rs7211029 were available in all 12 cohorts. The replication analyses showed consistency in the direction of the effect across most replication cohorts (Figure 3). The meta-analysis of the replication cohorts yielded significant evidence of association of rs9900428 (P-value=1.5 × 10−02), rs9907432 (P-value=7.1 × 10−03) and rs7211029 (P-value=2.5 × 10−03) with sleep latency (Table 2). The three SNPs are intronic to the RBFOX3 (also known as HRNBP3; Figure 1) and lie in the region with active regulatory elements (ENCODE) containing the H3K27ac mark and methylation marks. H3K27ac is an important enhancer mark that can distinguish between active and poised enhancer elements. Such enhancer elements are known to affect the expression of proximal genes and cluster near the genes they regulate.42 We investigated the three SNPs further using the methylome data of RS. The three SNPs were methylome-wide significantly associated (P-value=8.1 × 10−9, FDR=0.004) with the CpG site: cg16185152 in the RBFOX3 gene. We attempted to investigate the effect of methylation on gene expression, there was, however, only one probe for the RBFOX3 gene in the HumanHT-12_V4_Illumina 450K RNA expression array, which was removed in the quality control.

Forest plot for the three SNPs at chromosome 17q25. On the left, the populations including the populations in the GWAS and the replication phase. The boxes represent the precision and horizontal lines representing the confidence intervals. Pooled estimate is the effect estimate from the meta-analysis of all cohorts. The horizontal axis represents the scale of effects.
A gene network analysis of RBFOX3 using gene network tool (http://genenetwork.nl:8080/GeneNetwork/) shows strongest co-expression with the hippocalcin gene HPCA followed by SNCB, CABP1, JPH3, CPLX2, GABRA6, GABRD, NRXN3, RBFOX1, RTN4R, CNTN2 and WSCD2 among others (Supplementary Figure S5). A functional prediction of the gene showed involvement in the biological processes of synaptic functioning, membrane depolarization, gamma-aminobutyric acid (GABA) signaling and nervous system development (Supplementary Tables S5 and S6) and in dopamine, serotonin and glutamate neurotransmitters release cycle (Supplementary Table S7). RBFOX3 is expressed most strongly in various parts of the brain (brain, P-value=1.4 × 10−316; central nervous system, P-value=7.6 × 10−321; cerebral cortex, P-value=1.0 × 10−174) including the hypothalamus (P-value=9.7 × 10−10, AUC=0.96) (Supplementary Table S8), the locale of the SCN and a central part of the circadian clock.
None of the three SNPs showed strong association with sleep duration or mid-sleep, which is an indicator of the chronotype of an individual, Supplementary Table S9.
Discussion
In this first large-scale GWAS of sleep latency, we report the association of novel variants located in the gene RBFOX3 with sleep latency. Our gene discovery phase consisted of a sample of 4242 individuals from seven European populations, where sleep latency was assessed according to a common protocol. With this sample we discovered a cluster of three borderline genome-wide significant SNPs that were intronic to the gene RBFOX3. The replication of the three SNPs in up to 30 377 individuals from 12 independent cohorts showed significant association of the three variants and consistency in the direction of the effect estimates across most cohorts. A functional prediction of RBFOX3 based on a gene network analysis suggests significant involvement in the release cycle of neurotransmitters including GABA and various monoamines that are core to the human circadian clock, thus supporting the involvement of RBFOX3 with sleep latency.
The RBFOX3 gene (also called Fox-3, Hrnbp3, Neun) is located on the long arm of chromosome 17 (17q25). It belongs to the Fox-1 family of genes and shows high homology to RBFOX1 (also called Fox-1, A2bp1, Hrnbp1) and RBFOX2 (also called Fox-2, Rbm9, Hrnbp2). RBFOX3 is a relatively new member of this family and was recently identified to code for neuronal nuclei (NeuN) protein.43 The Fox proteins are a highly conserved family of tissue-specific splicing regulators.44 Although RBFOX1 is expressed in neurons, muscles and heart, and RBFOX2 in ovary, whole embryo, neurons and muscles,45, 46 the expression of RBFOX3 was detected exclusively in the post-mitotic regions of embryonic mouse brain.44, 47 RBFOX3 is believed to have a role in neuron-specific alternative splicing.43 Alternative splicing occurs most frequently in the brain,48, 49 presumably to generate large numbers of neuronal cell types and to support their diverse functions.43 A recent analysis of rodent SCN (the biological clock) anatomy using antibody against NeuN protein shows that RBFOX3 has a unique distribution which is limited to a particular sector of the SCN.50 Mutations in the FOX-1 genes lead to severe neuro-developmental phenotypes exhibiting mental retardation, epilepsy and autism spectrum disorder.51, 52, 53, 54 Further, a very recent study on patients with developmental delay detected a translocation disrupting the intron 2 of the RBFOX3 gene.55 Interestingly, this patient additionally had sleeping difficulties.
Gene-network analysis of RBFOX3 showed strong co-expression with genes involved in calcium channel activity and GABA signaling. GABA-ergic sleep neurons of the ventro-lateral pre-optic nucleus are activated by the circadian clock and adenosine, which progressively accumulates in the brain during waking. In turn these sleep-active neurons begin to inhibit the wake-active neurons via the neurotransmitter GABA.56 GABA-A receptors are the site of action of a number of important pharmacologic agents including barbiturates, benzodiazepines (sleep inducing drugs) and ethanol.57, 58 Polymorphisms in GABA-A receptors have been associated with insomnia.59 Functional prediction based on gene network analysis shows significant involvement of RBFOX3 in the release cycle of various neurotransmitters including dopamine, serotonin and glutamate, GABA-A receptor activation and Ras-activation upon Ca2+ influx through the NMDA receptor. Photic information is communicated to the molecular clockworks by release of glutamate from retino-hypothalamic nerve terminals and stimulation of glutamate receptors on SCN neurons.60 Glutamate stimulation is followed by intra-cellular increases of cyclic adenosine monophosphate and Ca2+ and activation of a Ras-dependent signal cascade in the circadian clockwork. Non-photic signaling to the core subdivision of the SCN is conveyed through two major pathways including GABA-containing neurons derived from the thalamus and serotonin or 5-hydroxytryptamine-containing neurons derived from the midbrain. Serotonergic input to the SCN shifts the timing of the clock.60
Our study shows strong evidence of association of sleep latency with RBFOX3. The fact that the association signal was stronger in the discovery sample compared with the replication sample, even though the replication sample was sixfold larger, may be explained by the differences in phenotyping as well as by different cohort characteristics (eg, age). Notably, all discovery cohorts were European, whereas the replication cohorts were drawn additionally from North America and Asia; it is possible that socio-cultural factors may have influenced sleep latency or how sleep latency was reported across cohorts. For instance, the phenotyping and quality control for the discovery cohorts was synchronized and centralized; all cohorts were assessed with MCTQ, which, unlike other such instruments, assesses information separately for free days and working days. Our GWAS was based on sleep latency on free days, as sleep latency on workdays is heavily influenced by daily professional activities. Unfortunately, this distinction was not available for most of the replication cohorts. Moreover, exclusions in the GWAS discovery cohorts were based not only on sleep medication use but also on other drugs that are known to influence sleep. Sleep medication or for that matter any medication that has a sleep-inducing effect reduces sleep latency,24 thereby introducing a potential bias on genetic studies of sleep latency. We also removed shift-workers and those using alarm clocks from the analysis in the discovery phase, which was not done in most replication cohorts. All of these factors likely affected the results in the replication phase. Nevertheless, despite a small effect size, the direction of association signal was consistent in most replication cohorts indicating the robustness of our finding. Furthermore, gene functional prediction and network analysis support the association of RBFOX3 variants with sleep latency. The predicted functioning of RBFOX3, including neurotransmitters’ release cycle and GABA-receptor activation strongly implicates a chronobiological explanation. However, further tests of association with various sleep disorders and functional analyses will provide a better insight into the relationship between RBFOX3 and sleep.
References
Krauchi K, Wirz-Justice A : Circadian clues to sleep onset mechanisms. Neuropsychopharmacology 2001; 25: S92–S96.
Cirelli C : The genetic and molecular regulation of sleep: from fruit flies to humans. Nat Rev Neurosci 2009; 10: 549–560.
Hastings M : The brain, circadian rhythms, and clock genes. BMJ 1998; 317: 1704–1707.
Hastings MH : Central clocking. Trends Neurosci 1997; 20: 459–464.
Baekeland F, Hoy P : Reported vs recorded sleep characteristics. Arch Gen Psychiatry 1971; 24: 548–551.
Hoch CC, Reynolds CF 3rd, Kupfer DJ, Berman SR, Houck PR, Stack JA : Empirical note: self-report versus recorded sleep in healthy seniors. Psychophysiology 1987; 24: 293–299.
Lehnkering H, Siegmund R : Influence of chronotype, season, and sex of subject on sleep behavior of young adults. Chronobiol Int 2007; 24: 875–888.
Geisler P, Tracik F, Cronlein T et al: The influence of age and sex on sleep latency in the MSLT-30—a normative study. Sleep 2006; 29: 687–692.
Krishnan V, Collop NA : Gender differences in sleep disorders. Curr Opin Pulm Med 2006; 12: 383–389.
Bonnet MH, Webb WB : The return to sleep. Biol Psychol 1979; 8: 225–233.
Afaghi A, O'Connor H, Chow CM : High-glycemic-index carbohydrate meals shorten sleep onset. Am J Clin Nutr 2007; 85: 426–430.
Nixon GM, Thompson JM, Han DY et al: Falling asleep: the determinants of sleep latency. Arch Dis Child 2009; 94: 686–689.
Gupta R, Dahiya S, Bhatia MS : Effect of depression on sleep: qualitative or quantitative? Indian J Psychiatry 2009; 51: 117–121.
Blader JC, Koplewicz HS, Abikoff H, Foley C : Sleep problems of elementary school children. A community survey. Arch Pediatr Adolesc Med 1997; 151: 473–480.
Eddy M, Walbroehl GS : Insomnia. Am Fam Physician 1999; 59: 1911–1916–1918.
Weitzman ED, Czeisler CA, Coleman RM et al: Delayed sleep phase syndrome. A chronobiological disorder with sleep-onset insomnia. Arch Gen Psychiatry 1981; 38: 737–746.
Freedman RR : EEG power spectra in sleep-onset insomnia. Electroencephalogr Clin Neurophysiol 1986; 63: 408–413.
van den Heuvel CJ, Lushington K : Chronobiology and insomnia: pathophysiology and treatment of circadian rhythm sleep disorders. Expert Rev Neurother 2002; 2: 249–260.
Touchette E, Petit D, Seguin JR, Boivin M, Tremblay RE, Montplaisir JY : Associations between sleep duration patterns and behavioral/cognitive functioning at school entry. Sleep 2007; 30: 1213–1219.
Heath AC, Kendler KS, Eaves LJ, Martin NG : Evidence for genetic influences on sleep disturbance and sleep pattern in twins. Sleep 1990; 13: 318–335.
Klei L, Reitz P, Miller M et al: Heritability of morningness-eveningness and self-report sleep measures in a family-based sample of 521 hutterites. Chronobiol Int 2005; 22: 1041–1054.
Partinen M, Kaprio J, Koskenvuo M, Putkonen P, Langinvainio H : Genetic and environmental determination of human sleep. Sleep 1983; 6: 179–185.
Kantermann T, Juda M, Merrow M, Roenneberg T : The human circadian clock's seasonal adjustment is disrupted by daylight saving time. Curr Biol 2007; 17: 1996–2000.
DeMartinis NA, Winokur A : Effects of psychiatric medications on sleep and sleep disorders. CNS Neurol Disord Drug Targets 2007; 6: 17–29.
Wittmann M, Dinich J, Merrow M, Roenneberg T : Social jetlag: misalignment of biological and social time. Chronobiol Int 2006; 23: 497–509.
Li Y, Abecasis GR : Mach 1.0: rapid haplotype reconstruction and missing genotype inference. Am J Hum Genet 2006; S79: 2290.
Li Y, Willer C, Sanna S, Abecasis G : Genotype imputation. Annu Rev Genomics Hum Genet 2009; 10: 387–406.
Howie BN, Donnelly P, Marchini J : A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 2009; 5: e1000529.
Marchini J, Howie B, Myers S, McVean G, Donnelly P : A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 2007; 39: 906–913.
Almasy L, Blangero J : Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet 1998; 62: 1198–1211.
Liu F, Kirichenko A, Axenovich TI, van Duijn CM, Aulchenko YS : An approach for cutting large and complex pedigrees for linkage analysis. Eur J Hum Genet 2008; 16: 854–860.
Aulchenko YS, Ripke S, Isaacs A, van Duijn CM : GenABEL: an R library for genome-wide association analysis. Bioinformatics 2007; 23: 1294–1296.
Amin N, van Duijn CM, Aulchenko YS : A genomic background based method for association analysis in related individuals. PLoS One 2007; 2: e1274.
Aulchenko YS, Struchalin MV, van Duijn CM : ProbABEL package for genome-wide association analysis of imputed data. BMC Bioinformatics 2010; 11: 134.
Wellcome Trust : Case Control C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007; 447: 661–678.
Chen WM, Abecasis GR : Family-based association tests for genomewide association scans. Am J Hum Genet 2007; 81: 913–926.
Orwoll E, Blank JB, Barrett-Connor E et al: Design and baseline characteristics of the osteoporotic fractures in men (MrOS) study—a large observational study of the determinants of fracture in older men. Contemp Clin Trials 2005; 26: 569–585.
Zaykin DV : Optimally weighted Z-test is a powerful method for combining probabilities in meta-analysis. J Evol Biol 2011; 24: 1836–1841.
Fehrmann RS, Karjalainen JM, Krajewska M et al: Gene expression analysis identifies global gene dosage sensitivity in cancer. Nat Genet 2015; 47: 115–125.
Alter O, Brown PO, Botstein D : Singular value decomposition for genome-wide expression data processing and modeling. Proc Natl Acad Sci USA 2000; 97: 10101–10106.
Sherlock G : Analysis of large-scale gene expression data. Brief Bioinform 2001; 2: 350–362.
Creyghton MP, Cheng AW, Welstead GG et al: Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA 2010; 107: 21931–21936.
Kim KK, Adelstein RS, Kawamoto S : Identification of neuronal nuclei (NeuN) as Fox-3, a new member of the Fox-1 gene family of splicing factors. J Biol Chem 2009; 284: 31052–31061.
Dredge BK, Jensen KB : NeuN/Rbfox3 nuclear and cytoplasmic isoforms differentially regulate alternative splicing and nonsense-mediated decay of Rbfox2. PLoS One 2011; 6: e21585.
Kiehl TR, Shibata H, Vo T, Huynh DP, Pulst SM : Identification and expression of a mouse ortholog of A2BP1. Mamm Genome 2001; 12: 595–601.
Underwood JG, Boutz PL, Dougherty JD, Stoilov P, Black DL : Homologues of the Caenorhabditis elegans Fox-1 protein are neuronal splicing regulators in mammals. Mol Cell Biol 2005; 25: 10005–10016.
McKee AE, Minet E, Stern C, Riahi S, Stiles CD, Silver PA : A genome-wide in situ hybridization map of RNA-binding proteins reveals anatomically restricted expression in the developing mouse brain. BMC Dev Biol 2005; 5: 14.
Modrek B, Resch A, Grasso C, Lee C : Genome-wide detection of alternative splicing in expressed sequences of human genes. Nucleic Acids Res 2001; 29: 2850–2859.
Yeo G, Holste D, Kreiman G, Burge CB : Variation in alternative splicing across human tissues. Genome Biol 2004; 5: R74.
Morin LP, Hefton S, Studholme KM : Neurons identified by NeuN/Fox-3 immunoreactivity have a novel distribution in the hamster and mouse suprachiasmatic nucleus. Brain Res 2011; 1421: 44–51.
Bhalla K, Phillips HA, Crawford J et al: The de novo chromosome 16 translocations of two patients with abnormal phenotypes (mental retardation and epilepsy) disrupt the A2BP1 gene. J Hum Genet 2004; 49: 308–311.
Barnby G, Abbott A, Sykes N et al: Candidate-gene screening and association analysis at the autism-susceptibility locus on chromosome 16p: evidence of association at GRIN2A and ABAT. Am J Hum Genet 2005; 76: 950–966.
Martin CL, Duvall JA, Ilkin Y et al: Cytogenetic and molecular characterization of A2BP1/FOX1 as a candidate gene for autism. Am J Med Genet B Neuropsychiatr Genet 2007; 144B: 869–876.
Sebat J, Lakshmi B, Malhotra D et al: Strong association of de novo copy number mutations with autism. Science 2007; 316: 445–449.
Utami KH, Hillmer AM, Aksoy I et al: Detection of chromosomal breakpoints in patients with developmental delay and speech disorders. PLoS One 2014; 9: e90852.
Luppi PH : Neurochemical aspects of sleep regulation with specific focus on slow-wave sleep. World J Biol Psychiatry 2010; 11 (Suppl 1): 4–8.
Whiting PJ : The GABA-A receptor gene family: new targets for therapeutic intervention. Neurochem Int 1999; 34: 387–390.
Whiting PJ, Bonnert TP, McKernan RM et al: Molecular and functional diversity of the expanding GABA-A receptor gene family. Ann N Y Acad Sci 1999; 868: 645–653.
Buhr A, Bianchi MT, Baur R et al: Functional characterization of the new human GABA(A) receptor mutation beta3(R192H). Hum Genet 2002; 111: 154–160.
Charney DS, Nestler EJ : Neurobiology of mental illness. Oxford University Press: Oxford; New York, NY, USA, 2004.
Acknowledgements
We thank all the participants and staff of all the studies for their co-operation and contribution. We are grateful to all participants and their relatives, general practitioners and neurologists for their contributions and to P Veraart for her help in genealogy, Jeannette Vergeer for the supervision of the laboratory work and P Snijders for his help in data collection for the ERF study. We thank Pascal Arp, Mila Jhamai, Marijn Verkerk, Lizbeth Herrera and Marjolein Peters for their help in creating the GWAS database, and Karol Estrada and Maksim V Struchalin for their support in creation and analysis of imputed data for the Rotterdam Study. We are grateful to the study participants, the staff from the Rotterdam Study and the participating general practitioners and pharmacists. We thank Dr Fernando Rivadeneira, Dr Tobias A Knoch, Anis Abuseiris, Luc V de Zeeuw and Rob de Graaf for their help in creating GRIMP, and BigGRID, MediGRID and Services@MediGRID/D-Grid. We acknowledge the invaluable contributions of the recruitment team from the Croatian Centre for Global Health, University of Split, the administrative teams in Croatia and Edinburgh and the people of Split. Our work was supported by the FP6 programme EUCLOCK, the Dutch Science Foundation (the NWO), the Hersenstichting Nederland, the Rosalind Franklin Fellowships of the University of Groningen, targeted Financing from the Estonian Government, the European Union through the European Regional Development Fund in the frame of Centre of Excellence in Genomics, FP7 Projects ECOGENE, BBMRI, ENGAGE and OPENGENE, the Geestkracht programme of the Dutch Scientific Organization (ZON-MW) and matching funds from participating universities and mental health care organizations, the Genetic Association Information Network (GAIN) of the Foundation for the US National Institutes of Health, the EUROSPAN (European Special Populations Research Network) through the European Commission FP6 STRP grant, the Chief Scientist Office of the Scottish Government, the Royal Society, the Medical Research Council, Erasmus University, Erasmus MC, the Centre for Medical Systems Biology (CMSB1 and CMSB2) and the Netherlands Genomics Initiative (NGI) and ALBAN. We acknowledge the High Performance Computing Center of University of Tartu, for space and facilities to conduct part of the EGCUT analysis. We also acknowledge the Netherlands Organization for Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), The Ministry of Education, Culture and Science, the Ministry of Health, Welfare and Sports of the Netherlands, the European Commission (DG XII) and the Municipality of Rotterdam. This study was supported by grants from the Netherlands Organization for Scientific Research (NWO-VENI grant 916.10.135 to LF) and a Horizon Breakthrough grant from the Netherlands Genomics Initiative (grant 92519031 to LF). The research leading to these results has received funding from the European Community’s Health Seventh Framework Programme (FP7/2007–2013) under grant agreement 259867. Study-specific acknowledgments and the ethical statement are provided in the Supplementary text.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Dr Najaf Amin is supported by the Netherlands Brain Foundation (project number F2013(1)-28). Dr Gregory J Tranah was supported by NIA grant R01AG030474. Dr Henning Tiemeier was supported by the Vidi Grant of ZonMw (the Netherlands Organization for Health Research and Development, 2009-017.106.370). All other authors report no biomedical financial interests or potential conflicts of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Amin, N., Allebrandt, K., van der Spek, A. et al. Genetic variants in RBFOX3 are associated with sleep latency. Eur J Hum Genet 24, 1488–1495 (2016). https://doi.org/10.1038/ejhg.2016.31
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2016.31
This article is cited by
-
Genome-wide association in Drosophila identifies a role for Piezo and Proc-R in sleep latency
Scientific Reports (2024)
-
Genetics of circadian rhythms and sleep in human health and disease
Nature Reviews Genetics (2023)
-
The End of Snoring? Application of CRISPR/Cas9 Genome Editing for Sleep Disorders
Sleep and Vigilance (2018)
-
Genome-wide association analysis of insomnia complaints identifies risk genes and genetic overlap with psychiatric and metabolic traits
Nature Genetics (2017)
