
Abstract
Coeliac disease (CD) is a chronic immune-mediated disease triggered by the ingestion of gluten. It has an estimated prevalence of approximately 1% in European populations. Specific HLA-DQA1 and HLA-DQB1 alleles are established coeliac susceptibility genes and are required for the presentation of gliadin to the immune system resulting in damage to the intestinal mucosa. In the largest association analysis of CD to date, 39 non-HLA risk loci were identified, 13 of which were new, in a sample of 12 014 individuals with CD and 12 228 controls using the Immunochip genotyping platform. Including the HLA, this brings the total number of known CD loci to 40. We have replicated this study in an independent Irish CD case–control population of 425 CD and 453 controls using the Immunochip platform. Using a binomial sign test, we show that the direction of the effects of previously described risk alleles were highly correlated with those reported in the Irish population, (P=2.2 × 10−16). Using the Polygene Risk Score (PRS) approach, we estimated that up to 35% of the genetic variance could be explained by loci present on the Immunochip (P=9 × 10−75). When this is limited to non-HLA loci, we explain a maximum of 4.5% of the genetic variance (P=3.6 × 10−18). Finally, we performed a meta-analysis of our data with the previous reports, identifying two further loci harbouring the ZNF335 and NIFA genes which now exceed genome-wide significance, taking the total number of CD susceptibility loci to 42.
Similar content being viewed by others


Introduction
Coeliac disease (CD) is a complex and chronic immune-mediated disease affecting the small intestine with a prevalence of approximately 1% in the European population.1, 2 Coeliac patients are sensitive to the ingestion of gliadin and related proteins, which initiate an immune response ultimately leading to villous atrophy and flattening of the intestinal mucosa. CD is a polygenic disorder with probandwise concordance rates in monozygotic twins of over 80% compared with 17% in dizygotic twins leading to estimates of heritability in excess of 80%.3, 4 The HLA locus is the most important inherited susceptibility factor, variously estimated to account for up to 40% of the genetic variance.5, 6, 7, 8 The vast majority of CD patients carry either HLA-DQ2 or DQ8 heterodimers, which are expressed on antigen-presenting cells and present gliadin to T cells. However, environmental and non-HLA loci also contribute to susceptibility because CD-associated HLA-DQ molecules are not alone sufficient to cause CD susceptibility, which is evident from the fact that they are common in the general population (usually reported as 30% or greater), whereas only a small proportion of carriers will ever develop CD.9
A number of genome-wide association studies (GWAS) have been carried out to identify additional CD susceptibility loci. The first study was performed in a UK sample of 778 patients and 1422 controls10 and led to the unequivocal identification of the first non-HLA risk locus in CD, the IL2/IL21 region of chromosome 4 (odds ratio (OR)=0.63, CI (0.57–0.71)). A subsequent GWAS in a larger multinational sample brought the number of established non-HLA risk loci to 26.11, 12 Early GWAS arrays, however, failed to adequately capture much of the genetic variation of disease-associated loci, making it difficult to identify the location of the true risk variant. Furthermore, the lack of uniformity in marker selection between different arrays made comparisons with other diseases difficult. Consequently, the Immunochip was developed, a low cost Illumina Infinium genotyping array containing 196 524 common and rare polymorphisms at loci implicated in GWAS of 12 major autoimmune and inflammatory diseases including CD.13 The dense, targeted, genotyping afforded by this array was designed to allow improved signal localisation and cross disease comparison.
Trynka and colleagues,14 using the Immunochip in a study of 12 014 CD patients and 12 228 controls from seven geographic regions, reported 57 independent CD association signals across 39 non-HLA loci. These consisted of loci meeting genome-wide significance (GWS; P<5 × 10−8) in their data and/or in previous CD GWAS and replication data sets. Notably, this included 13 loci not previously associated with CD, bringing the total number of CD risk loci to 40 including the HLA. Similar to other complex diseases, the individual effect size for most of these observed non-HLA variants is small (OR 1.12–1.36) and together they explain only a small proportion of disease risk.
It has been shown that by combining the loci of small effect, including those that do not achieve GWS, it is possible to explain a greater proportion of the variance in complex traits. One approach to capturing this genetic liability is the polygenic risk score (PRS) described by Purcell and colleagues.15 The PRS combines the effects of multiple common variants and has been successfully used to examine the influence of risk alleles en masse to disease susceptibility in a range of complex disorders such as psychiatric illness,16 multiple sclerosis17 and cardiovascular disease.18 In essence, the PRS is a risk score for an individual calculated from the cumulative burden of risk alleles carried by that individual; these risk alleles are then weighted according to their observed effect size on disease (OR) to come up with the final PRS. In CD, similar approaches to PRS have been applied by Romanos and colleagues,19 who developed a CD genetic risk model by combining HLA plus the 57 non-HLA SNP associated loci, and Abraham and colleagues,20 who developed genomic risk scores for CD based on approximately 250 SNPs.
In this analysis, we first set out to perform an association study using the Immunochip array in an independent Irish population of 425 CD and 453 controls and to present these new data in the context of previously identified association signals. Secondly, we performed meta-analyses of these new data combined with those reported by Trynka and colleagues14 to test whether this led to the discovery of any additional GWS loci. Thirdly, in order to examine the polygenic contribution across studies, we derived PRSs for HLA and non-HLA CD loci identified and weighted according to the findings from the Trynka et al14 association study (discovery set) and calculated the proportion of variance explained for the Irish study (estimated via Nagelkerke’s pseudo R2). Finally, we investigated the discriminatory capacity of the PRS derived from our study to distinguish case status in this sample.
Materials and methods
Sample description
All individuals are Caucasian and have self-reported Irish ancestry as defined by four grandparents of exclusively Irish origin. Genomic DNA was extracted from peripheral blood samples. CD were assigned on the basis of positive serology to antibody-based tests (anti-tTG and anti-EMA) followed by examination of the histological appearance of small intestinal biopsy samples and noting clinical response to a gluten-free diet. Controls were an anonymous unselected (ie not screened for CD) healthy sample of blood donors from the general population provided by the TCD/IMM population DNA biobank. Written and informed consent was obtained from all subjects, and the study was approved by the Research Ethics Committees of St James’s Hospital Dublin and all other contributing centers.
Genotyping and genotyping quality control
Genotyping was performed by using the Illumina Infinium High-Density array (Illumina Inc., San Diego, CA, USA) at the UCD Academic Centre on Rare Diseases (ACoRD). Genotypes were called using the Illumina GenomeStudio software using the cluster set of 172 242 autosome/x-chromosome variants from the Trynka et al14 study (Immuno_BeadChip_11419691_B.bpm, NCBI build 36 (hg18)).
All quality control of the genotype data was performed using PLINK v1.07.21 Additional calculations and visualisations were performed in STATA v13 using self-authored scripts. Individual genotyping samples were excluded based on per-sample call rate (missing >5%), excessive autosomal heterozygosity (>3SD from sample mean) and evidence of relatedness (up to and including second degree relatives). The population was restricted to a proxy-North Western European ancestry subset as defined by similarity to the HAPMAP CEPH reference dataset. Population outliers were identified by multi-dimensional scaling and individuals >3SD from the CEPH mean component score for the first two principal components were removed. Individual SNPs were removed from the association analysis for low call rate (<95%) deviation from Hardy–Weinberg equilibrium in controls (P<1 × 10−5), or low minor allele frequency (<0.01). Following quality control, 425 CD cases and 453 controls genotyped across 143 074 markers were available for analysis.
Genetic association
Association analysis was performed in PLINK v1.0721 using logistic regression adjusting for gender and 10 population covariates (principal components generated from the multi-dimensional scaling routine in PLINK). Raw genotype intensity plots for all markers described in Table 1 were visually inspected to confirm cluster separation and genotyping quality. These data have been made publically available at the GWAS Central database22 at the URL(http://www.gwascentral.org/study/HGVST1830).
Meta-analysis
Meta-analyses of the findings from our data and those of Trynka et al14 were performed using METAL.23 Prior to analysis, SNPs were corrected for strand inconsistencies; any SNPs where strand ambiguity was possible (A/T or C/G) were excluded from analysis. Allele frequencies were compared between the Irish and UK controls from the Trynka et al14 dataset and were found to be highly concordant (r2=0.996). Studies were combined using an inverse variance weighted approach combining the association signal (P-values). We also calculated I2 as a measure of heterogeneity.
Sign test
To examine whether the observed direction of effect in our sample was concordant with markers previously identified as GWS by Trynka et al,14 a binomial sign-test was performed. To limit the effect of linkage disequilibrium on these data, linkage-independent SNPs were identified in the discovery set by applying the —clump flag in PLINK. Briefly, sequentially from the most associated marker (P<5 × 10−8), all additional SNPs within a 1 Mb window that were correlated with it at an r2>0.2 were excluded. This left us with 280 LD-independent GWS SNPs in total.
PRS analyses
Using the Trynka et al14 association study as the discovery set, polygene scores were calculated for the Irish study individuals using the —score flag in PLINK v1.07. In total, we calculated eight PRS limited to SNPs that met an association threshold in the discovery analyses. These thresholds were P<5 × 10−8, P<0.01, P<0.05, P<0.1, P<0.2, P<0.3, P<0.4 and P<0.5. All analyses were calculated on the putative risk allele and a relative weight was applied to each allele as the log OR of the association signal observed in the discovery analyses. Individual PRS were then calculated as the sum of all risk variants carried by that individual weighted for the effect size. To limit the effect of linkage disequilibrium on these data, linkage-independent SNPs were identified in the discovery set as described above.
To examine the predictive strength of the derived PRS in our sample, a nested logistic regression was performed. All scores were converted to a scale of 0–1 and individual SNP missingness was corrected by mean imputation from available SNPs genotyped in the individual. Two nested logistic regression models were applied; (i) case status predicted by score, gender and population covariates and (ii) case status predicted by gender and population covariates.
The variance explained by the score (Vm) was calculated as the difference in Nagelkerke’s pseudo R2 between the two models. In addition to performing score analyses for all associated markers, analyses on SNPs within the HLA/MHC locus only and SNPs not within the HLA/MHC locus (non-HLA) were also carried out. In these analyses, the model tested included the alternate set as a covariate, such that for HLA markers we examined (i) case status predicted by score_HLA, score_non-HLA, gender and population covariates and (ii) case status predicted by score_non-HLA, gender and population covariates.
The Student’s t-test was used to test for significance of the differences in risk score between groups and we also calculated the Receiver Operator Characteristic for the scores in the Irish study. All statistical analyses were performed in STATA v13.
Results
Genetic association
As expected, a highly significant association was observed within the HLA region in our study of 425 Irish coeliac patients and 453 population controls. The frequency of HLA-DQ2.5 carriers (inferred by rs2187668 chr6:hg18:g.32713862C>T genotype) was 88% in Irish individuals with celiac disease versus 29% of controls, consistent with previous findings.24 The genomic inflation factor lambda (λ) was calculated as 1.12. The strongest association was observed for rs3891175 which maps to HLA-DQB1 (P=1.9 × 10−32, OR=6.22 (95% CI 4.6–8.4)). A summary of the top LD independent findings is given in Supplementary Table 1. Outside of the MHC/HLA region, no additional SNPs achieved GWS (Supplementary Table 2). Manhattan and QQ plots for SNPs outside of the HLA are shown in Figure 1. Supplementary Figure 1a shows Manhattan and QQ plots for all SNPs and Supplementary Figure 1b QQ plots for the null set of SNPs present on the chip that are not associated with autoimmune diseases.

QQ (left) and Manhattan plot (right) of −log10 (P values) for association for all SNPs on the Immunochip to CD in the Irish sample excluding the HLA/MHC locus (chr6:20–40 Mb).
Replication
Trynka et al14 reported 57 GWS, LD-independent, non-HLA CD association signals that reached levels of GWS or had been identified as such in earlier CD GWAS and replication data sets.11, 12 Fifty-six of these SNPs passed QC in our dataset (imm_21_44453549 was omitted because of low call rate), and of these, we observed 5 SNPs at P<0.005 and 6 SNPs below or at the nominal association level (P<0.05) (Table 1). On the basis of the null hypothesis, we would expect to observe 0 at P<0.005 and less than 3 at P<0.05. This represents strong evidence for replication and provides additional support for IL2/IL21 (Pirish <1 × 10−4), SCHIP1, IL12A, PTPN2, SH2B3, ETS1, TAGAP (Pirish <1 × 10−3), PVT1, DDX6 (Pirish <1 × 10−2) and RUNX3, RGS1 (Pirish <0.05) as risk loci for CD.
Moreover, by performing a meta-analysis combining the data of Trynka et al14 and this study, the association signal was strengthened at several known loci (Supplementary Figure 2), and two additional loci were identified which surpassed GWS levels in the combined analysis; NFIA (rs6691768 chr1:hg18:g.61564451G>A,Pcombined=2.2 × 10−8) and ZNF335 (rs6032606 chr20:hg18:g44029614C>G,Pcombined=4.81 × 10−8) (Supplementary Table 3).
To test the consistency of the direction of associations with those previously reported, a binomial sign test was used. There was highly significant evidence of concordance in direction of effect between the Trynka et al14 and the Irish studies. Of 280 GWS SNPs, 218 (78%) show effect in the same direction (Pr (K>=218)=2.2 × 10−16). Notably, this trend was maintained when analyses were restricted to the 57 GWS, LD-independent, non-HLA CD association signals, where 43 of 56 (77%) show the same direction of effect (Pr (K>=43)=5.6 × 10−05) (Supplementary Table 4).
PRS analyses
PRS were calculated at eight association thresholds in our sample (<5 × 10−8, P<0.01, P<0.05, P<0.1, P<0.2, P<0.3, P<0.4 and P<0.5). The analysis was applied to three sets of SNPs; all markers, HLA only and non-HLA. When all markers are included, higher PRS were found to be significantly associated with CD case status at each of the eight thresholds (Table 2). This was also true when we examined HLA SNPs only. For the non-HLA SNP set, higher PRS were significantly associated with coeliac case status at the thresholds P<5 × 10−8, P<0.01, P<0.05 and P<0.1 (see Supplementary Figures (i)-(xvi)).
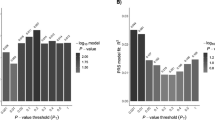
Figure 2 shows the proportion of variance explained by PRS at each of the different thresholds. Up to 35% of the variance was explained by the PRS when all markers were considered (SNPs threshold P<0.05; Student’s t test, P=9.23 × 10−75). Furthermore, up to 4.5% of the variance could be explained by the non-HLA SNPs alone (SNPs threshold P<0.01; P=3.6 × 10−18). Up to 31% of the variance was explained by the HLA SNPs alone (SNPs threshold P<0.1; P =2 × 10−70).

Variance explained in an Irish Coeliac cohort by PRS derived from LD-independent SNPs from Trynka et al.14 Red represents the percentage variance explained (R2) when all markers are analysed (red) and for the non-HLA markers (teal). Shading indicates the proportion of total SNPs included in the model.
For each threshold, we also estimated the area under the receiver operating characteristic curve (AUC) (see Supplementary Table 5). For all markers, the highest AUC was 0.83 (SNPs threshold P <0.05) (Figure 3a); for non-HLA markers, the AUC was 0.67 (SNPs threshold P<0.01) (Figure 3b); and for HLA only, the highest AUC was 0.82 (SNPs threshold P<0.5).

Receiver operator characteristic (ROC) curves and AUCs for the risk scores at (a) including all markers under the 0.05 threshold and (b) for the non-HLA markers at the 0.01 threshold.
Discussion
Recent association studies have added substantially to our understanding of the genetic complexity of CD, making it one of the best understood complex diseases. However, there is still a low probability of replicating GWS results, so it is important that disease-associated loci for complex diseases should be replicated in independent populations.25
In this study, we sought to replicate in an independent Irish case–control sample, findings from a recent large Immunochip analysis carried out by Trynka and colleagues.14 We defined replicated SNPs as those showing nominally significant association (P<0.05) and a consistent direction of effect. Trynka and colleagues14 reported 57 independent non-HLA association signals for CD that had reached GWS, either in their dataset, or earlier genome-wide studies. We found evidence of replication at 11 of these SNPs, which was almost four times greater than would be expected by chance at the P<0.05 level. Genes at these loci include IL2-IL21, PTPN2, SCHIP1-IL12A, SH2B3, ETS1, TAGAP, PVT1, TREH-DDX6, RUNX3 and RGS1. Furthermore, there was highly significant evidence of concordance in direction of effect for the majority (77%) of SNPs that were GWS in the Trynka et al14 study in our data (both HLA and non-HLA SNPs). This included 12 of the new loci identified by Trynka et al14 that had not been previously implicated in CD, adding further support for these loci as true coeliac susceptibility loci.
Increasing the sample size in association studies has been shown to have more impact in terms of SNP discovery than improving array coverage even with imputation.26 More risk loci for CD and related diseases remain to be detected. With even the current large sample sizes, many true signals of association fall short of the threshold for GWS. In the largest recent GWAS (of anthropometrics,27 Crohn’s disease28 and schizophrenia), increases in sample size have yielded a non-linear increase in the identified GWS loci. In schizophrenia, for example, an increase in sample participants from approximately 3000 to 6000 to 9000 and more recently 35 000 resulted in the discovery of 1, 2, 6 and 108 GWS loci, respectively.29
As demonstrated in this study, the modest increase in sample size brought about by combining the Irish and the existing Immunochip dataset added sufficient power to raise an additional two loci beyond the GWS threshold; rs6691768 (PTrynka=5.3 × 10−8; Pcombined=2.28 × 10−8) and rs6032606 (PTrynka=8.25 × 10−8; Pcombined=4.81 × 10−8).
rs6691768 is an intronic SNP located in the NFIA gene, which is a member of the NF1 (nuclear factor 1) family of transcription factors. It had been identified by Dubois et al11 as a locus with suggestive evidence for CD in an earlier GWAS (PDubois combined 1.19 × 10−7). NFIA has been most extensively studied in neurological tissue although there is evidence that it plays an important role in myeloid lineages.30
rs6032606 is a missense SNP located in the gene ZNF335 which has not previously been specifically implicated in CD and is not strongly associated with other immune mediated diseases, although located in an extended region identified as a risk locus for Crohn’s disease, multiple sclerosis and rheumatoid arthritis (ImmunoBase; www.immunocase.org). ZNF335 (also known as NIF-1) was originally shown to modulate nuclear hormone receptor coactivator activity, thus indirectly affecting the function of a wide variety of nuclear ligand-bound and other transcription factors including Fos, Jun and NFkB.31, 32 More recently, it has been shown to be a component of the trithorax chromatin remodelling complex,33 with H3K4 (and perhaps other31) methyltransferase activity, and its deletion results in embryonic lethality.33 It is a binding partner of another transcriptional coregulator with chromatin-remodelling activity HCFC131 (HCF1). HCFC1 is also encoded in a CD susceptibility locus on Xq28.14 Thus, ZNF335 can profoundly influence gene expression and in combination with HCFC1, implicates chromatin remodeling as a molecular mechanism in CD pathogenesis.
In a PRS analysis using the Trynka and colleagues14 GWAS summary statistics as a discovery dataset, we examined the collective influence of risk alleles on disease susceptibility and assessed the predictive power of these SNPs present on the Immunochip. Similar approaches have already been undertaken in CD19, 20 but not solely using the Immunochip. By simultaneously considering variants in the Immunochip data en masse, we observed strong evidence that regardless of P-value threshold used, alleles associated with CD from the earlier studies can explain a significant proportion of the variance in our CD cohort. Increase in the liability is not limited to markers reaching the standard benchmarks of association, for example, GWS or P<0.05, but also extends to those with P<0.5, indicating there are more disease-associated markers to be found amongst these non-GWS SNPs.
The highest proportion of variance in disease status explained by the score in our dataset was 35% when all markers are included (P<0.05 threshold) and up to 4.5% excluding the HLA locus (P<0.01). The latter finding is in accordance with previous observations in CD and other complex conditions including schizophrenia15 and multiple sclerosis,17 in which common non-HLA variants have been found to explain approximately 3% of the variance in disease risk. In contrast with other studies, however, the proportion of variance explained decreased when more liberal discovery thresholds (P>0.1) were used to calculate the polygenic scores, indicating that the signal was being diluted by the addition of more biologically non-relevant SNPs. The fact that the Immunochip is not representative of genome-wide variation but rather a defined subset of loci selected on the basis of their association with inflammatory diseases may also influence this observation.
Our findings are consistent with that of Abraham and colleagues20 who, using a machine learning approach, generated a PRS based on multiple SNPs that explains 30–35% of disease variance when applied across several CD cohorts. Their analysis was performed predominately on datasets genotyped on the Ilumina Infinium array platform but also included a dataset genotyped on the Immunochip (however, only SNPs in common to both were included – approximately 4% overlap). Their data showed a predictive power of 0.86–0.9 (AUC), which was moderately higher than that reported by Ramonos et al19 who assessed the predictive power of just the 57 non-HLA susceptibility variants combined with HLA genotypes (AUC=0.854). The highest AUC in our dataset using SNPs present on the Immunochip was 0.83 (SNPs threshold P<0.05) including the HLA, and 0.67 (SNPs threshold P<0.01) with non-HLA markers only.
It is currently estimated that up to 54% of the heritability of CD can be explained through a combination of HLA and non-HLA genetic factors.34 This still leaves a substantial proportion of ‘missing heritability’ to be uncovered, and to date, there is little evidence that rare variants are contributing substantially to the heritability of CD.35 This polygene study provides further evidence that common variants are contributing to the disease. However, PRS are limited in that only narrow sense heritability (ie a purely additive model of association) is examined. This method does not take into account the proportion of variation that is attributable to broad sense heritability, that is, additive, non-additive, multiplicative and epistatic interactions. Recent studies have found evidence of epistasis contributing to both psoriasis36 and multiple sclerosis37 disease risk, indicating that it plays an important role in risk to other autoimmune diseases but may be difficult to detect owing to the lack of power or other technical considerations.
One of the biggest advantages of the Immunochip is that it costs a fraction of standard GWAS chips and is applicable to other immune diseases. This study shows it has added value as a predictor of CD case status and it will be instructive to see whether this also applies to other related diseases. Studies of inflammatory bowel disease indicates this to be likely, where AUCs of 0.86 and 0.83 were predicted for Crohn’s disease and ulcerative colitis, respectively.38 PRS is a flexible method that, used in conjunction with low-cost genotyping platforms similar to the Immunochip, could represent a valuable and cost-efficient tool with clinical application in assessing the risk of developing these diseases.
Our small sample size is a limitation of this study and meant that we had insufficient power to observe associations of small effect (<4% power to detect an association at P <5 × 10−8 (minor allele frequency 0.5, OR <1.36). Even so, we observe PRS scores that are sufficiently different between cases and controls to predict case status (AUC 0.83) in line with previous reports.
In conclusion, we have replicated in an independent Irish CD sample a number of the findings from a larger Immunochip study confirming associations at 11 loci. The direction of effect of GWS signals were highly concordant and our findings show that the Immunochip can be used to predict CD case status accurately using a polygene score method. Finally, by combining our data with earlier Immunochip studies, we have identified the regions encoding NFIA and ZNF335 as GWS CD susceptibility loci.
References
Abadie V, Sollid LM, Barreiro LB, Jabri B : Integration of genetic and immunological insights into a model of celiac disease pathogenesis. Ann Rev Immunol 2011; 29: 493–525.
Sollid LM, Jabri B : Triggers and drivers of autoimmunity: lessons from coeliac disease. Nat Rev Immunol 2013; 13: 294–302.
Greco L, Romino R, Coto I et al: The first large population based twin study of coeliac disease. Gut 2002; 50: 624–628.
Nisticò L, Fagnani C, Coto I et al: Concordance, disease progression, and heritability of coeliac disease in Italian twins. Gut 2006; 55: 803–808.
Bevan S, Popat S, Braegger CP et al: Contribution of the MHC region to the familial risk of coeliac disease. J Med Genet 1999; 36: 687–690.
Karell K, Louka AS, Moodie SJ et al: HLA types in celiac disease patients not carrying the DQA1*05-DQB1*02 (DQ2) heterodimer: results from the European Genetics Cluster on Celiac Disease. Hum Immunol 2003; 64: 469–477.
Margaritte-Jeannin P, Babron MC, Bourgey M et al: HLA-DQ relative risks for coeliac disease in European populations: a study of the European Genetics Cluster on Coeliac Disease. Tissue Antigens 2004; 63: 562–567.
Trynka G, Wijmenga C, van Heel DA : A genetic perspective on coeliac disease. Trends Mol Med 2010; 16: 537–550.
Megiorni F, Mora B, Bonamico M et al: HLA-DQ and risk gradient for celiac disease. Hum Immunol 2009; 70: 55–59.
van Heel DA, Franke L, Hunt KA et al: A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat Genet 2007; 39: 827–829.
Dubois PCA, Trynka G, Franke L et al: Multiple common variants for celiac disease influencing immune gene expression. Nat Genet 2010; 42: 295–302.
Hunt KA, Zhernakova A, Turner G et al: Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet 2008; 40: 395–402.
Parkes M, Cortes A, van Heel DA, Brown MA : Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat Rev Genet 2013; 14: 661–673.
Trynka G, Hunt KA, Bockett NA et al: Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat Genet 2011; 43: 1193–1201.
Purcell SM, Wray NR, Stone JL et al. The International Schizophrenia Consortium: Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009; 460: 748–752.
Cross-Disorder Group of the Psychiatric Genomics Consortium: Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 2013; 381: 1371–1379.
Bush WS, Sawcer SJ, de Jager PL et al. The International Multiple Sclerosis Genetics Consortium: Evidence for polygenic susceptibility to multiple sclerosis—the shape of things to come. Am J Hum Genet 2010; 86: 621–625.
Simonson MA, Wills AG, Keller MC, McQueen MB : Recent methods for polygenic analysis of genome-wide data implicate an important effect of common variants on cardiovascular disease risk. BMC Med Genet 2011; 12: 146.
Romanos J, Rosén A, Kumar V et al: Improving coeliac disease risk prediction by testing non-HLA variants additional to HLA variants. Gut 2014; 63: 415–422.
Abraham G, Tye-Din JA, Bhalala OG, Kowalczyk A, Zobel J, Inouye M : Accurate and robust genomic prediction of celiac disease using statistical learning. PLoS Genet 2014; 10: e1004137.
Purcell S, Neale B, Todd-Brown K et al: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Beck T, Hastings RK, Gollapudi S, Free RC, Brookes AJ : GWAS Central: a comprehensive resource for the comparison and interrogation of genome-wide association studies. Eur J Hum Genet 2014; 22: 949–952.
Willer CJ, Li Y, Abecasis GR : METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010; 26: 2190–2191.
Sollid LM : Molecular basis of celiac disease. Ann Rev Immunol 2000; 18: 53–81.
Liu Y-J, Papasian CJ, Liu J-F, Hamilton J, Deng H-W : Is replication the gold standard for validating genome-wide association findings? PLoS ONE 2008; 3: e4037.
Lindquist KJ, Jorgenson E, Hoffmann TJ, Witte JS : The impact of improved microarray coverage and larger sample sizes on future genome-wide association studies. Genet Epidemiol 2013; 37: 383–392.
Wood AR, Esko T, Yang J et al: Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet 2014; 46: 1173–1186, advance online publication.
Jostins L, Ripke S, Weersma RK et al: Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012; 491: 119–124.
Schizophrenia Working Group of the Psychiatric Genomics C: Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014; 511: 421–427.
Starnes LM, Sorrentino A, Ferracin M et al: A transcriptome-wide approach reveals the key contribution of NFI-A in promoting erythroid differentiation of human CD34+ progenitors and CML cells. Leukemia 2010; 24: 1220–1223.
Mahajan MA, Murray A, Samuels HH : NRC-interacting factor 1 is a novel cotransducer that interacts with and regulates the activity of the nuclear hormone receptor coactivator NRC. Mol Cell Biol 2002; 22: 6883–6894.
Garapaty S, Xu C-F, Trojer P, Mahajan MA, Neubert TA, Samuels HH : Identification and characterization of a novel nuclear protein complex involved in nuclear hormone receptor-mediated gene regulation. J Biol Chem 2009; 284: 7542–7552.
Yang Yawei J, Baltus Andrew E, Mathew Rebecca S et al: Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation. Cell 2012; 151: 1097–1112.
Kumar V, Wijmenga C, Withoff S : From genome-wide association studies to disease mechanisms: celiac disease as a model for autoimmune diseases. Semin Immunopathol 2012; 34: 567–580.
Hunt KA, Mistry V, Bockett NA et al: Negligible impact of rare autoimmune-locus coding-region variants on missing heritability. Nature 2013; 498: 232–235.
Strange A, Capon F, Spencer CC et al. Genetic Analysis of Psoriasis Consortium & the Wellcome Trust Case Control Consortium 2: A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet 2010; 42: 985–990.
Lincoln MR, Ramagopalan SV, Chao MJ et al: Epistasis among HLA-DRB1, HLA-DQA1, and HLA-DQB1 loci determines multiple sclerosis susceptibility. Proc Natl Acad Sci USA 2009; 106: 7542–7547.
Wei Z, Wang W, Bradfield J et al: Large sample size, wide variant spectrum, and advanced machine-learning technique boost risk prediction for inflammatory bowel disease. Am J Hum Genet 2013; 92: 1008–1012.
Acknowledgements
We acknowledge the input of the TCD Biobank, and Dr J McPartlin and M Cunneen, for access to DNA population samples. We thank all patients and unaffected individuals for providing samples for this study. The Dublin Centre for Clinical Research (funded by the HRB and Wellcome Trust) conducted much of the patient sample collection. We thank Dr David A van Heel for managed open access to coeliac disease Immunochip data. This work was funded by the Science Foundation Ireland (award 09/IN.1/B2640 to RMM). GT was funded by the Wellcome Trust Sanger Institute (WT098051).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Coleman, C., Quinn, E., Ryan, A. et al. Common polygenic variation in coeliac disease and confirmation of ZNF335 and NIFA as disease susceptibility loci. Eur J Hum Genet 24, 291–297 (2016). https://doi.org/10.1038/ejhg.2015.87
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.87
This article is cited by
-
Genetics of irritable bowel syndrome: shifting gear via biobank-scale studies
Nature Reviews Gastroenterology & Hepatology (2022)
-
Identification of genomic regions affecting production traits in pigs divergently selected for feed efficiency
Genetics Selection Evolution (2021)
-
Independent and cumulative coeliac disease-susceptibility loci are associated with distinct disease phenotypes
Journal of Human Genetics (2021)
-
Genetic architecture of type 1 diabetes with low genetic risk score informed by 41 unreported loci
Communications Biology (2021)
-
Replication study and meta-analysis indicate a suggestive association of RUNX3 locus with primary biliary cholangitis
Immunogenetics (2020)
