
Abstract
Pathogenic mitochondrial DNA (mtDNA) point mutations are associated with a wide range of clinical phenotypes, often involving multiple organ systems. We report two patients with isolated myopathy owing to novel mt-tRNAAla variants. Muscle biopsy revealed extensive histopathological findings including cytochrome c oxidase (COX)-deficient fibres. Pyrosequencing confirmed mtDNA heteroplasmy for both mutations (m.5631G>A and m.5610G>A) whilst single-muscle fibre segregation studies (revealing statistically significant higher mutation loads in COX-deficient fibres than in COX-positive fibres), hierarchical mutation segregation within patient tissues and decreased steady-state mt-tRNAAla levels all provide compelling evidence of pathogenicity. Interestingly, both patients showed very high-mutation levels in all tissues, inferring that the threshold for impairment of oxidative phosphorylation, as evidenced by COX deficiency, appears to be extremely high for these mt-tRNAAla variants. Previously described mt-tRNAAla mutations are also associated with a pure myopathic phenotype and demonstrate very high mtDNA heteroplasmy thresholds, inferring at least some genotype:phenotype correlation for mutations within this particular mt-tRNA gene.
Similar content being viewed by others


Introduction
Mitochondrial DNA (mtDNA) disorders are associated with a wide range of different clinical phenotypes, from mild to severe.1 Mutations affecting mitochondrial (mt)-tRNA genes are prevalent amongst adults and usually associated with multisystemic disease presentations;2 isolated organ involvement is rarely observed. Furthermore, it is unusual for mutations in one specific mt-tRNA gene to associate with a unique clinical phenotype, although several mutations in mt-tRNALeu(UUR) and mt-tRNAIle are linked to MELAS and mitochondrial cardiomyopathy respectively.3
The overwhelming majority of pathogenic mtDNA mutations are present in a heteroplasmic state, the level of mtDNA mutation within a cell or tissue required to exceed a critical threshold to cause a disease phenotype.4 This threshold level varies for each mutation and tissue and is dependant on several factors including OXPHOS metabolism.5, 6 Here we report two patients, both presented with isolated myopathy, with novel heteroplasmic mutations in the mt-tRNAAla gene exhibiting high thresholds for disease expression.
Patients and methods
Patient 1
Patient 1 is a young lady who presented at the age of 29 years with muscle weakness, initially involving her hands but spreading to her arms and legs progressively, resulting in her being non-ambulatory by the age of 40 years. Her mother is healthy with no history of muscle disease, whereas the patient has no children or siblings. She was noted to have a mild symmetrical ptosis but with no history of myoglobinuria, diabetes or seizures. Neurological examination revealed general floppy tetraparesis (MRC 3-4/5). Muscle tendon reflexes were weak. Sensory examination results, tone and Babinski reflexes were all normal. Electroencephalogram revealed no signs of increased cerebral excitability. Needle electromyogram of the biceps brachii muscle and the vastus medialis muscle revealed distinctive myopathic changes. Nerve conduction studies of the suralis nerve showed normal results. Thigh-MRI showed distinctive fatty degeneration of all muscle groups, equal on both sides. Cerebral MRI, cardiac MRI and audiogram were normal. Creatine kinase was slightly elevated (3.9 μmol/l; normal: <2.4 μmol/l). Resting lactate level was normal (2.0 mmol/l; normal: <2.8 mmol/l).
Patient 2
This lady presented at the age of 69 years with progressive weakness of the limb girdle muscles including proximal paresis. There is no known muscle disease or muscle weakness in the family. Ptosis, ophthalmoplegia or extramuscular mitochondrial symptoms have not been noticed. Neurological examination revealed dysarthria, a facies myopathica and proximal paresis (MRC 3-4/5). Sensory examination and Babinski reflexes were normal. Muscle tendon reflexes on the upper extremity were weak. Achilles deep-tendon reflexes were not obtainable on both sides. Patella deep-tendon reflexes were left accentuated obtainable. Needle electromyogram of the deltoideus muscle and the biceps brachii muscle revealed myopathic changes. Creatine kinase was slightly elevated (2.7 μmol/l; normal: <2.4 μmol/l), as was the resting lactate level (2.9 mmol/l; normal: <2.8 mmol/l).
Histopathology and molecular genetic studies
Standard histopathologic analysis of muscle biopsies of both patients was performed and the activities of respiratory chain complexes were determined spectrophotometrically.7 Total DNA from all available tissue (muscle, urinary epithelia, buccal epithelia, hair shafts and blood), including tissue from available maternal relatives, was extracted by standard procedures. Long-range PCR of muscle DNA was undertaken to detect large-scale mtDNA rearrangements,8 followed by sequencing of the entire mitochondrial genome in this tissue.9, 10 Analysis of mtDNA heteroplasmy was carried out by quantitative pyrosequencing including segregation studies within individual cytochrome c oxidase (COX)-positive and COX-deficient fibres.11 High-resolution northern blotting to assess mt-tRNAAla steady-state levels in both patients was performed as described.12
Results
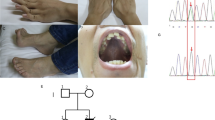
Muscle biopsy analysis revealed numerous COX-deficient (Patient 1: 33%, Patient 2: 40%) and ragged-red-fibres (Patient 1: 2%, Patient 2: 5%) in both patients. Biochemical analysis of muscle from Patient 1 showed decreased activities of respiratory chain complexes I, II/III and IV, and a compensatory increase in citrate synthase activity. Decreased activities of respiratory chain complexes I and IV, with a compensatory increase in citrate synthase activity, were noted in Patient 2. Long-range PCR failed to identify large-scale mtDNA deletions, prompting sequencing of the entire mitochondrial genome in muscle which revealed novel mt-tRNAAla (MTTA) gene mutations – m.5631G>A (ClinVar Reference SCV000196082: NC_012920.1) in Patient 1 and m.5610G>A (ClinVar Reference SCV000196083: NC_012920.1) in Patient 2 (Figure 1c). The highest m.5631G>A mutation load in Patient 1 was found in muscle (92% levels of mtDNA heteroplasmy), with lower levels present in blood (77%), hair shafts (77%), urinary epithelial sediment (69%) and buccal epithelial cells (44%). In Patient 2, the highest mutant load was detected also in muscle (91% levels of mtDNA heteroplasmy), followed by blood (87%). Levels of mtDNA heteroplasmy detected in additional family members are shown (Figures 2a and b).

Identification of novel mt-tRNAAla variants (a) Sequencing electropherogram (reverse sequence) demonstrating the heteroplasmic m.5631G>A transition detected in patient muscle. (b) Sequencing electropherogram demonstrating the heteroplasmic m.5610G>A transition detected in muscle. (c) Schematic representation of the mt-RNAAla cloverleaf structure, illustrating the position of the novel m.5631G>A and m.5610G>A variants and other reported mt-tRNAAla mutations. Phylogenetic conservation of the appropriate regions of the mt-tRNAAla gene sequence for both (d) m.5631G>A and (e) m.5610G>A indicates that both variants affect an evolutionary conserved residue.

Molecular genetic investigations of patients’ muscle with the novel m.5631G>A and m.5610G>A variants (a) Patient 1’s pedigree including mtDNA mutation heteroplasmy levels in the index case and her mother. (b) Family pedigree including mtDNA mutation heteroplasmy levels in the index case (Patient 2) and other family members. (c) Single-fibre PCR analysis of the m.5631G>A mutation segregates with a biochemical defect in individual COX-deficient muscle fibres. (d) Single-fibre PCR analysis of the m.5610G>A mutation segregates with a biochemical defect in individual COX-deficient muscle fibres. (e) Measurement of mt-tRNAAla steady-state levels in muscle showing dramatically decreased mt-tRNAAla steady-state levels in both patients.
Single-muscle fibre analysis of individual COX-positive and COX-deficient fibres of both cases revealed a statistically significant higher mutation load in COX-deficient fibres than in COX-positive fibres (Patient 1: COX-deficient fibres: 95.1% ±0.45 (n=18), COX-positive fibres: 83.8%±3.38 (n=22), P=0.005; Patient 2: COX-deficient fibres: 98.0%±0.46 (n=20), COX-positive fibres: 78.9%±4.81 (n=21), P=0.0002; Figures 2c and d), whilst the muscle from both patients showed dramatically decreased mt-tRNAAla steady-state levels compared with normal controls (Figure 2e).
Discussion
Histopathological findings of diagnostic muscle biopsies in both patients were characterised by numerous COX-deficient fibres and evidence of subsarcolemmal mitochondrial accumulation. This prompted us to thoroughly investigate the mitochondrial genome. The two novel heteroplasmic mt-tRNA point mutations identified, m.5631G>A and m.5610G>A, are both located in mt-tRNAAla and are unequivocally pathogenic according to accepted criteria published by Yarham and colleagues.13
Additional samples from maternally related members of both patients were available for investigation (Figures 2a and b). Interestingly, one daughter of Patient 2 had obviously higher mutation loads in blood and buccal cells than all other investigated family members, although well below the expected disease threshold for the m.5610G>A mutation. She is healthy, with no sign of muscle weakness although she declined detailed investigations such as assessment of CK levels and formal clinical examination.
mtDNA mutations are frequently associated with multisystemic diseases, although isolated organ involvement is rarely observed. Isolated myopathy owing to mt-tRNA mutations is a rare clinical presentation. Mutations were detected not only in the mt-tRNAAla gene but single cases are described with mutations in the mt-tRNAAsp, mt-tRNATrp, mt-tRNAPhe, mt-tRNAGln, mt-tRNALeu(CUN), mt-tRNAPro, mt-tRNAIle, mt-tRNASer(UCN) and mt-tRNALys genes. Mutations in the mt-tRNAAla gene all appear to be associated with isolated myopathy in contrast to mutations in these other mt-tRNA genes that are associated with different phenotypes including multisystemic presentations. In addition to the novel mutations presented here, there are four other previously reported examples of mt-tRNAAla mutations, two of which are associated with myopathy affecting the limbs (m.5591G>A and m.5650G>A) and two mutations (m.5628T>C and m.5636T>C) associated with a myopathy affecting additionally the extraocular and pharyngeal muscles (Table 1 and Figure 1c). The patient previously described with a m.5591G>A transition presented with pure myopathy, involving predominantly limb girdle muscles, myalgia, elevated CK levels and a severe combined respiratory chain enzyme defect.14 Follow-up analysis of urine samples from his two unaffected brothers revealed the presence of the m.5591G>A mutation (67% mutant load) in the younger brother, but not in the older brother.14 Initially, the younger brother presented with asymptomatic elevation of CK, later developing exercise-induced muscle weakness leading to proximal paresis at the age of 51 years. The patient reported with a maternally inherited m.5650G>A mutation in mt-tRNAAla suffered from an increasingly severe limb girdle myopathy.15 Two other reported cases showed a preference for skeletal muscle involvement; the m.5628T>C mutation was identified in a patient who presented with proximal paresis, episodic diplopia, external ophthalmoplegia and dysphagia,16 whilst the m.5636T>C mutation was identified in a patient who presented with fatigue, bilateral ptosis, external ophthalmoplegia and mild dysarthria.17
It is interesting that the threshold for impairment of oxidative phosphorylation, as evidenced by COX deficiency, appears to be extremely high for both novel mt-tRNAAla mutations. The previously described cases of mt-tRNAAla mutations associated with a pure myopathic phenotype (m.5591G>A and m.5650G>A) demonstrated similarly high-mutation thresholds, inferring at least a genotype: phenotype correlation for mutations within this particular mt-tRNA gene.
References
Taylor RW, Turnbull DM : Mitochondrial DNA mutations in human disease. Nat Rev Genet 2005; 6: 389–402.
Blakely EL, Yarham JW, Alston CL et al: Pathogenic mitochondrial tRNA point mutations: nine novel mutations affirm their importance as a cause of mitochondrial disease. Hum Mutat 2013; 34: 1260–1268.
Yarham JW, Elson JL, Blakely EL, McFarland R, Taylor RW : Mitochondrial tRNA mutations and disease. Wiley Interdiscip Rev RNA 2010; 1: 304–324.
DiMauro S, Moraes CT : Mitochondrial encephalomyopathies. Arch Neurol 1993; 50: 1197–1208.
Wong LJ : Diagnostic challenges of mitochondrial DNA disorders. Mitochondrion 2007; 7: 45–52.
Tuppen HA, Blakely EL, Turnbull DM, Taylor RW : Mitochondrial DNA mutations and human disease. Biochimica et Biophysica Acta 2010; 1797: 113–128.
Gellerich FN, Deschauer M, Chen Y, Muller T, Neudecker S, Zierz S : Mitochondrial respiratory rates and activities of respiratory chain complexes correlate linearly with heteroplasmy of deleted mtDNA without threshold and independently of deletion size. Biochimica et Biophysica Acta 2002; 1556: 41–52.
Deschauer M, Kiefer R, Blakely EL et al: A novel Twinkle gene mutation in autosomal dominant progressive external ophthalmoplegia. Neuromuscul Disord 2003; 13: 568–572.
He L, Chinnery PF, Durham SE et al: Detection and quantification of mitochondrial DNA deletions in individual cells by real-time PCR. Nucleic Acid Res 2002; 30: e68.
Tuppen H A, Hogan V E, He L et al: The p.M292T NDUFS2 mutation causes complex I-deficient Leigh syndrome in multiple families. Brain 2010; 133: 2952–2963.
White HE, Durston VJ, Seller A, Fratter C, Harvey JF, Cross NC : Accurate detection and quantitation of heteroplasmic mitochondrial point mutations by pyrosequencing. Genet Test 2005; 9: 190–199.
Taylor RW, Giordano C, Davidson MM et al: A homoplasmic mitochondrial transfer ribonucleic acid mutation as a cause of maternally inherited hypertrophic cardiomyopathy. J Am Coll Cardiol 2003; 41: 1786–1796.
Yarham JW, McFarland R, Taylor RW, Elson JL : A proposed consensus panel of organisms for determining evolutionary conservation of mt-tRNA point mutations. Mitochondrion 2012; 12: 533–538.
Swalwell H, Deschauer M, Hartl H et al: Pure myopathy associated with a novel mitochondrial tRNA gene mutation. Neurology 2006; 66: 447–449.
McFarland R, Swalwell H, Blakely EL et al: The m.5650G>A mitochondrial tRNAAla mutation is pathogenic and causes a phenotype of pure myopathy. Neuromuscul Disord 2008; 18: 63–67.
Spagnolo M, Tomelleri G, Vattemi G, Filosto M, Rizzuto N, Tonin P : A new mutation in the mitochondrial tRNAAla gene in a patient with ophthalmoplegia and dysphagia. Neuromuscul Disord 2001; 11: 481–484.
Pinos T, Marotta M, Gallardo E et al: A novel mutation in the mitochondrial tRNAAla gene (m.5636T>C) in a patient with progressive external ophthalmoplegia. Mitochondrion 2011; 11: 228–233.
Acknowledgements
We acknowledge the technical assistance of Kathleen Zietz, Thekla Wangemann and Gavin Falkous. DL, PRJ, SZ and MD are members of the German mitoNET funded by the German Ministry of Education and Research. RWT is supported by a Wellcome Trust Strategic Award (096919Z/11/Z), the Medical Research Council (UK) Centre for Translational Muscle Disease Research (G0601943), The Lily Foundation and the UK NHS Specialist Commissioners which funds the ‘Rare Mitochondrial Disorders of Adults and Children’ Diagnostic Service in Newcastle upon Tyne (http://www.mitoresearch.org.uk/).
AUTHOR CONTRIBUTIONS
Experimental work, preparation of manuscript: DL and PRJ; experimental work: KS, SAH, HALT and KB; interpretation of the data and critical review: ELB and SZ; critical review: CB; concept, supervision, interpretation of the data and critical review: MD and RWT.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Lehmann, D., Schubert, K., Joshi, P. et al. Pathogenic mitochondrial mt-tRNAAla variants are uniquely associated with isolated myopathy. Eur J Hum Genet 23, 1735–1738 (2015). https://doi.org/10.1038/ejhg.2015.73
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.73
