
Abstract
The expression gradient of the morphogen Sonic Hedgehog (SHH) is crucial in establishing the number and the identity of the digits during anteroposterior patterning of the limb. Its anterior ectopic expression is responsible for preaxial polydactyly (PPD). Most of these malformations are due to the gain-of-function of the Zone of Polarizing Activity Regulatory Sequence, the only limb-specific enhancer of SHH known to date. We report a family affected with a novel condition associating PPD and hypertrichosis of the upper back, following an autosomal dominant mode of inheritance. This phenotype is consistent with deregulation of SHH expression during limb and follicle development. In affected members, we identified a 2 kb deletion located ~240 kb upstream from the SHH promoter. The deleted sequence is capable of repressing the transcriptional activity of the SHH promoter in vitro, consistent with a silencer activity. We hypothesize that the deletion of this silencer could be responsible for SHH deregulation during development, leading to a PPD-hypertrichosis phenotype.
Similar content being viewed by others


Introduction
Anteroposterior patterning of the limb is initiated during early development to assign the number and the identity of the digits. The zone of polarizing activity (ZPA) is a mesenchymatous region of the posterior margin and is recognized as center for this signaling pathway. ZPA cells express the secreted morphogen Sonic Hedgehog (SHH, OMIM#600725), encoding a signal protein. Its gradient of expression determines the differentiation of the digits.1 The anterior ectopic expression of SHH is responsible for congenital limb malformations, in particular preaxial polydactyly (PPD).2, 3, 4, 5, 6 PPD has an incidence of ~1/2000 births. Most PPDs are due to the gain-of-function (by pathogenic variant or copy number gain) of a SHH cis-regulatory element. This element, the ZPA regulatory sequence (ZRS, OMIM#605522), has an enhancer activity specific for the developing bud (for review, see ref. 7). This enhancer is located ~1 Mb upstream of SHH (7q36). This distance can slightly differ among vertebrate species while the ordering of genes and regulatory elements is highly conserved, therefore probably having a crucial role in SHH regulation. The existence of other cis-regulatory elements of SHH specific for limb development is likely, as numerous PPD families linked to the 7q36 region do not have a ZRS alteration.8, 9
We studied a large family affected with autosomal dominant PPD. Limb malformations were associated with hypertrichosis affecting the upper back. This phenotypic association has not, to our knowledge, been previously reported and we hypothesize that this could be due to SHH deregulation. We first confirmed the linkage at the 7q36 locus in this family, performing haplotyping with SNP-arrays. ZRS sequencing and copy number analysis was normal. Sequencing of ECRs among the candidate locus did not reveal any pathogenic variant. We then identified a 2 kb deletion at long distance upstream of SHH, comprising a silencer. Deletion of this regulatory element could lead to anterior overexpression of SHH, therefore causing PPD. Given the role of SHH during follicle morphogenesis and growth,10, 11, 12 it is likely that its deregulation is also responsible for the hypertrichosis in this family.
Material and methods
Patients
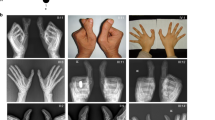
Through the Clinical Genetics department of Lille University Hospital, France, we recruited a large family of European ancestry comprising 25 index cases in five generations (pedigree, Figure 1) presenting with PPD and upper back hypertrichosis, following an autosomal dominant mode of inheritance. Phenotypic expressivity was variable among individuals, ranging from triphalangeal thumbs, duplicated thumbs, preaxial extra ray to syndactyly between digits I and II (Table 1,Figure 2, Supplementary Figures 1 and 2). All affected members presented with hirsutism, starting as a low posterior hairline and spreading down to the middle of the back (Table 1, Figure 2,Supplementary Figure 3). Written patient consents were obtained prior to the analyses. Standard karyotype was normal in some affected individuals that were tested.

Pedigree of the family affected with autosomal dominant preaxial polydactyly and upper back hypertrichosis. Individuals available for phenotyping and genotyping are indicated with an asterisk.

Clinical and radiological findings in patients affected with preaxial polydactyly and hypertrichosis. (a) Hand malformations are variable, ranging from triphalangeal thumb, biphalangeal partly duplicated thumb, preaxial extra ray to syndactyly between digits I and II. (b) Foot malformations are variable, ranging from large to duplicated hallux, potentially associated with syndactyly between the first and second rays. (c) Pictures showing upper back hypertrichosis, starting as a low posterior hairline and spreading down to the middle of the back. More clinical pictures are available in Supplementary Figures 1–3.
Array-CGH
We performed array-CGH 44 K (Agilent Technologies, Courtaboeuf, France) in one affected member of the family (IV-21), to search for copy number variation, according to the manufacturer's protocol. Male genomic DNA was used as a reference in sex-match hybridization and results were analyzed with the CGH analytics software.
ZRS screening
ZRS Sanger sequencing was performed in affected members of the family using three couples of primers (primers’ sequences are provided in Supplementary Table 1) to study the whole ZRS region (chr7.hg19:g.156,583,690-156,584,790). The sequence was compared with the reference sequence (Ref Seq, hg19, NC_000007.14) using SeqScape software.
To assess ZRS copy number variations, we performed a quantitative PCR assay with two primers’ couples (primers’ sequences are provided in Supplementary Table 1) in affected individuals. The reaction was performed with the SYBR Green technology according to the manufacturer’s protocol (Applied Biosystems). Quantification of the target sequences was normalized to an assay from RPPH1 gene, and the relative copy number was determined on the basis of the comparative ΔΔCt method using a normal control DNA as the calibrator.
Haplotyping by SNP-arrays
We performed SNP-arrays (CytoSNP-12 v2.1, Illumina, San Diego, USA) in 16 family members (I-1, II-1, II-2, II-3, II-3’s partner, III-2, III-3, III-9, III-12, III-20, IV-3, IV-4, IV-5, IV-7, IV-19 and IV-21). Samples were processed according to the manufacturer’s protocol, and results were analyzed using Illumina GenomeStudio software. Lod-scores were calculated using Merlin software13 with the following parameters: autosomal dominant model, 100% penetrance, frequency of disease 0.0001, rate of de novo mutation 0.0001 in both sexes.
Evolutionary conserved regions study
To identify the evolutionary conserved regions (ECRs) comprised in the candidate locus, we performed human genomic sequence alignment with the orthologous sequences of mice and chicken using the browser http://ecrbrowser.dcode.org with the following parameters: minimal ECR length 350 bp, minimal ECR similarity 77% (threshold defined in14). The ECRs were sequenced in an affected individual using a custom capture design SureSelect (Agilent Technologies) on the MiSeq (Illumina), according to the manufacturers’ protocols. The design was performed with the SureDesign software and variant calling with the SureCall software (Agilent Technologies).
Plasmid constructs
Constructs were generated by inserting target sequence into the pLS_Prom_SHH vector (pLightSwitch_Prom_SHH vector, #S716462, Active Motif, La Hulpe, Belgium). This plasmid contains a minimal SHH promoter and the Renilla luciferase reporter gene. The 2026 bp sequence of interest was obtained by PCR on genomic DNA and cloned upstream of the SHH promoter in the pLS_Prom_SHH vector using the MluI restriction site (primers’ sequences are provided in Supplementary Table 1). Two constructs were generated, with forward (pLS_Prom_SHH_2026F) and reverse pLS_Prom_SHH_2026R) orientation of the 2026 bp sequence.
As controls, we used the pLS_Prom_SHH vector and the pLS_EmptyProm (pLightSwitch_EmptyProm, #S790005, Active Motif). The latter does not contain any promoter upstream of the Renilla luciferase reporter gene and was used as a measure of background signal in the experiment.
To normalize with the transfection efficiency, we performed co-transfections with the pGL3 Promoter Vector (#E1761, Promega, Charbonnieres, France) that contains a SV40 promoter and the Firefly luciferase reporter gene.
Cell culture and luciferase assay
Cells of the human colon adenocarcinoma cell line Caco2 were maintained in 20% FBS/MEM containing 110 mg/l sodium pyruvate, 0.1 mM non-essential amino acids, 1% glutamine and 0.5% of penicillin–streptomycin. Cells at 60–70% confluence were transfected with 0.4 μg Renilla luciferase reporter constructs and 50 ng Firefly luciferase reporter plasmid using the Effectene transfection reagent (Qiagen, Courtaboeuf, France) in six-well plates, according to the manufacturer’s protocol. After 48 h of transfection, cells were washed twice with phosphate-buffered saline and lysed with passive lysis buffer (Promega). Renilla and Firefly luciferase reporter activities were assessed with the Dual-Luciferase Reporter Assay (Promega) according to the manufacturer’s protocol, using the Mithras LB 940 Multimode Microplate Reader (Berthold Technologies, Thoiry, France). The Renilla/Firefly luciferase activity ratio for each construct was compared with the pLS_Prom_SHH construct. The transfections were performed in triplicate, three times.
Results
ZRS study is normal in PPD-hypertrichosis
ZRS Sanger sequencing in affected individuals of the family revealed no pathogenic variant and quantitative PCR assay showed two copies of the limb-specific enhancer (data not shown).
PPD-hypertrichosis is linked to the 7q36 locus
No copy number variation was identified in the family by array-CGH 44 K (mean resolution 150 kb). To map the PPD-hypertrichosis condition, we performed whole genome haplotyping by SNP-arrays in 16 individuals from the family. Results showed linkage to a single locus at 7q36 (Lod-score>3). There was no other locus in the genome with a positive Lod-score (Supplementary Figure 4). The candidate region spans 2.1 Mb between rs2305944 and rs4101 (chr7.hg19:g.155809123_157947033), containing seven genes, seven non-coding RNAs and one micro-RNA (Figure 3). It is located upstream of the SHH gene and encompasses its limb-specific enhancer, the ZRS.

(a) Locus for PPD-hypertrichosis identified by SNP-arrays haplotyping (delimited by the dotted line). The region spans 2.1 Mb at 7q36 (chr7.hg19:g.155809123_157947033). It is located upstream of the SHH gene and encompasses the ZRS. (b) SNP array profile for the linkage region showing the heterozygous deletion of rs4296934 (arrow). (c) Sanger sequencing of the deletion breakpoint. Identification of a heterozygous 2026 bp deletion in the gene desert upstream SHH segregating in affected individuals.
The candidate locus contains several ECRs
The phenotypic association of PPD and hypertrichosis is highly suggestive of SHH deregulation. Given that the ZRS study (sequencing and quantitative PCR) was normal in this family, we hypothesized that another SHH regulatory element located at 7q36 could be disrupted. In this hypothesis, we identified 66 ECRs (including the ZRS) in the candidate locus of 2.1 Mb (Supplementary Table 2). Some of these have already been described and studied in PPD families.8, 9 Sequencing of these regions in one individual affected with PPD-hypertrichosis (IV-21) revealed no pathogenic variant.
Identification of a 2 kb deletion segregating with the phenotype
The data generated by SNP-arrays in the candidate region of 2.1 Mb were reanalysed for copy number variation. We identified a heterozygous deletion of rs4296934 located in the gene desert upstream of SHH, at ~240 kb from its promoter (Figure 3). This deletion was confirmed by long-range PCR-sequencing (Figure 3) and spans 2026 bp (chr7.hg19:g.155845627_155847652). This deletion segregates with the phenotype in the family with complete penetrance. The deletion was absent from 100 control chromosomes. The data were submitted in the Decipher database (https://decipher.sanger.ac.uk, #296668). No identical copy number variation was reported in this database. We therefore hypothesize that this deletion could be responsible for the PPD-hypertrichosis phenotype by the deregulation of SHH expression.
Structure of the 2 kb sequence
After mapping the PPD-hypertrichosis phenotype to the 7q36 locus, we identified a 2026 bp region deleted in the family. Inter-species alignments show that the wild-type sequence is highly conserved between human and rhesus primate, and partially conserved in mouse, dog, chicken and zebrafish (Supplementary Figure 4). In the less conserved parts of the region, three repetitive elements are located: one DNA repeat (Mer112) of 113 bp, one simple repeat of 76 bp and one Alu sequence of 298 bp (Supplementary Figure 5). Besides, the 2026 bp sequence contains numerous predicted binding sites for transcription factors (MatInspector, Genomatix). We selected the sites for which the matrix similarity was over 95% and filtered the transcription factors for which less than three binding sites were predicted. With these criteria, 28 transcription factors have three or more predicted binding sites on the sequence (Supplementary Table 3). Regarding these data, we hypothesize that this region could possibly contain a regulatory element.
The 2 kb deletion contains a cis-regulatory element
To elucidate a possible regulatory effect of the 2026 bp element, a functional study was initiated in cell cultures. The 2026 bp sequence was subcloned into the expression vector pLS_Prom_SHH. As the human colon adenocarcinoma Caco2 cell line expresses SHH, we therefore performed transfections in these cells with the construct containing the 2026 bp sequence in forward (pLS_Prom_SHH_2026F) and reverse orientation (pLS_Prom_SHH_2026R). Both constructs show an approximately twofold decrease of the relative reporter activity, as compared with the control vector without regulatory element pLS_Prom_SHH (Figure 4). Thus, the 2026 bp sequence contains an element capable of repressing the transcriptional activity of the SHH promoter in cis, consistent with a silencer activity.

Functional study of the 2026 bp element. Relative luciferase expression was examined in Caco2 cells using a reporter construct pLS_Prom_SHH_2026 containing the 2026 bp sequence that is deleted in patients affected with PPD-hypertrichosis, in forward (F) or reverse (R) orientation. Reporter activity was normalized to the pLS_Prom_SHH control vector that do not contains any regulatory element upstream of the SHH promoter. In presence of the 2026 bp sequence and in absence of the promoter, the reporter activity is significantly reduced (****P<0.0001). Error bars represent the SD obtained from three independent experiments performed in triplicates.
Discussion
We report a large family affected with PPD and upper back hypertrichosis, with autosomal dominant inheritance and variable expressivity. To our knowledge, this phenotypic association has never been described before. The main differential diagnoses are non-syndromic PPDs (Supplementary Table 4), as the hypertrichosis of the back can be overlooked, especially in males. Non-syndromic PPDs are caused by gain-of-function mutations or copy number gains of the ZRS, a limb-specific enhancer of SHH. Besides, some cases of non-syndromic PPDs have been reported in association with GLI3 mutations (OMIM#165240).15, 16 GLI3 is responsible for Greig cephalopolysyndactyly syndrome (GCPS, OMIM#175700) and Pallister–Hall Syndrome (PHS, OMIM#146510). GCPS can be very variable but usually presents with crossed polysyndactyly (PPD of lower limbs and postaxial polydactyly of upper limbs), macrocephaly, corpus callosum agenesis and sometimes craniosynostosis. In PHS, the polydactyly is usually meso-axial or postaxial and is associated with hypothalamic hamartoma. It is debated if GLI3 could effectively be responsible for non-syndromic polydactyly or if this is would only be the milder end of GCPS or PHS spectrum.17 Hypertrichosis has not been reported in GLI3-associated syndromes, except for some cases harboring 7p13 microdeletions involving GLI3.18 This feature is therefore likely due to the contiguous gene deletion.
Given the central role of SHH in the anteroposterior polarization of the limb and in the follicle morphogenesis,10, 11, 12 we hypothesized that the PPD-hypertrichosis phenotype is consistent with a deregulation of SHH expression. No pathogenic variant or copy number variation was identified in the ZRS, which is the only limb-specific enhancer of SHH known to date. Nevertheless, the regulatory architecture of SHH necessary for limb development seems incompletely defined so far. Other limb-specific cis-regulatory elements of SHH are likely, as numerous PPD families with linkage at 7q36 do not present with ZRS alteration.8, 9 The identification of a 4–6 kb deletion between ZRS and SHH in acheiropodia, which phenotype is consistent with the loss of SHH expression in the developing limb, is a further clue.
In this PPD-hypertrichosis family, linkage at the 7q36 locus upstream of the SHH gene was confirmed by SNP-arrays haplotyping. We identified the loss of a 2026 bp region in the gene desert 240 kb from the SHH promoter in our patients. This deleted region is partially conserved among species and absent from the Database of Genomic Variants and in 100 controls tested. Transfection experiments showed that the deleted sequence contains an element capable of repressing the transcriptional activity of the SHH promoter, consistent with a silencer activity. Therefore, we hypothesize that the deletion of this novel cis-regulatory element in our patients could explain SHH overexpression during limb development, leading to PPD. However, our present data do not allow us to rule out that another molecular mechanism could be responsible for the phenotype: this deletion may lead to disrupted interactions between SHH and other regulatory elements; alternatively, a variant in a potential unknown regulatory element within the candidate region may remain undetected. Furthermore, in vitro studies of the regulation of genes involved in limb development are difficult to design because of the lack of relevant cell lines available. For these transfection experiments, we chose to use the human colon adenocarcinoma cell line Caco2 because it expresses SHH and has been used before successfully to study the limb-specific ZRS enhancer.19 The existence of multiple regulatory elements in addition to the ZRS is likely, similarly to the regulatory clusters orchestrating the spatiotemporal pattern of SHH expression necessary to the development of the central nervous system20, 21 and the epithelial linings.22 The regulatory element identified may interact with the SHH promoter located at long distance by formation of a chromosome loop, similarly to the ZRS enhancer.23 The SHH spatial and temporal expression pattern may be directed by the assemblage of these two (or more) autonomous regulatory activities.
In silico analysis of the candidate sequence shows numerous predicted binding sites for transcription factors. Among these, four transcription factors are good candidates to explain the regulatory activity of our sequence, given the literature data. ETS and HAND transcription factor families have, respectively, six and four predicted binding sites on the sequence that is deleted in the PPD-hypertrichosis family. These factors, in particular ETS1/ETV4/ETV5 (OMIM#164720, 600711, 601600) and HAND2 (OMIM#602407), are known to have central roles in the SHH spatial pattern during limb development via their direct binding to the ZRS.24, 25 Furthermore, variants of ETV4/5 binding sites on the ZRS are responsible for its ectopic activity leading to PPD.25 To our knowledge, these transcription factor families have not been involved in follicle morphogenesis or growth.
Binding sites for GATA and NKX family factors are over-represented on the 2 kb sequence and especially along its more evolutionary conserved part, with, respectively, 16 and 33 predicted sites, suggesting a ‘homotypic clustering’. The latter, corresponding to the enrichment of multiple binding sites for the same transcription factor, is a common feature of cis-regulatory elements in vertebrate and invertebrate.26 These two families of transcription factors are known to act synergistically to regulate tissue-specific gene expression during development.27 This co-regulation activity has been well described for GATA4-NKX2.5 in cardiogenesis28, 29, 30 and for GATA6-NKX2.1 in lung development.27, 31 To our knowledge, no major role in limb development has been described for NKX factors so far. Interestingly, it has recently been demonstrated that Gata4 and Gata6 are differentially expressed in the anterior mesenchyme of the developing limb buds in mice.32 Gata6 represses ectopic expression of Shh in the anterior region of the limb bud by directly binding to the ZRS. The limb bud-specific Gata6 deletion results in ectopic expression of Shh and its target genes in the anterior margin, therefore resulting in PPD. A simultaneous deletion of Gata6 and Shh rescues the phenotype. Conversely, forced-expression of Gata6 throughout the limb bud downregulates Shh, resulting in a decreased number of digits. Therefore, Gata transcription factors are crucial negative regulators of ectopic Shh expression during limb development. Furthermore, these factors contribute to transcriptional regulation by facilitating chromosome looping, thereby mediating long-range gene regulation.33 Given these observations, we hypothesize that GATA transcription factors, and in particular GATA6, could bind to the regulatory element deleted in the PPD-hypertrichosis patients, conferring its silencing activity.
Given the role of SHH during follicle morphogenesis and growth, it is likely that its deregulation is also responsible for the hypertrichosis in the family. Wnt/β-catenin signaling relayed through Shh and Bmp signals is the principal regulatory mechanism of hair follicle development.34 Studies in mice showed that follicle morphogenesis and hair growth is altered when Shh is either downregulated10, 11, 12 or overexpressed.35 This pathway is deregulated by knock-out of Gata3 in mice, the animals showing abnormal hair formation.36 Therefore, GATA transcription factors are candidates for interacting with the regulatory element identified, responsible for deregulation of SHH in the developing limb and hair follicle.
Chromatin immunoprecipitation experiments may be conducted to identify which transcription factors are involved in the regulatory activity of the 2 kb element described. Similarly to the ZRS, the interaction between this cis-regulatory element and the SHH promoter located at a 240 kb distance could occur by chromosomal looping. Chromosome conformation capture experiments may be performed to validate this hypothesis. Unfortunately, in vivo experiments would be difficult to design for two reasons: first, the limited choice of a relevant animal model for the two phenotypes (ie, polydactyly and hypertrichosis); second, the weak inter-species conservation of the potential regulatory element restricting even more this choice. Consequently, study of this newly suspected SHH limb regulatory element in other families affected with PPD-hypertrichosis may be the best approach to confirm our findings. Nevertheless, this phenotype has never been described before and is therefore presumed to be an extremely rare condition in humans. Of note, screening of the 2 kb element by sequencing and quantitative PCR of 20 patients affected with isolated PPD (for whom ZRS sequence and copy number variation have been previously ruled out) revealed no molecular variation (data not shown).
References
Riddle RD, Johnson RL, Laufer E, Tabin C : Sonic hedgehog mediates the polarizing activity of the ZPA. Cell 1993; 75: 1401–1416.
Heus HC, Hing A, van Baren MJ et al: A physical and transcriptional map of the preaxial polydactyly locus on chromosome 7q36. Genomics 1999; 57: 342–351.
Heutink P, Zguricas J, van Oosterhout L et al: The gene for triphalangeal thumb maps to the subtelomeric region of chromosome 7q. Nat Genet 1994; 6: 287–292.
Hing AV, Helms C, Slaugh R et al: Linkage of preaxial polydactyly type 2 to 7q36. Am J Med Genet 1995; 58: 128–135.
Tsukurov O, Boehmer A, Flynn J et al: A complex bilateral polysyndactyly disease locus maps to chromosome 7q36. Nat Genet 1994; 6: 282–286.
Zguricas J, Heus H, Morales-Peralta E et al: Clinical and genetic studies on 12 preaxial polydactyly families and refinement of the localisation of the gene responsible to a 1.9 cM region on chromosome 7q36. J Med Genet 1999; 36: 32–40.
VanderMeer JE, Ahituv N : cis-regulatory mutations are a genetic cause of human limb malformations. Dev Dyn 2011; 240: 920–930.
Albuisson J, Isidor B, Giraud M et al: Identification of two novel mutations in Shh long-range regulator associated with familial pre-axial polydactyly. Clin Genet 2011; 79: 371–377.
Li H, Wang CY, Wang JX et al: Mutation analysis of a large Chinese pedigree with congenital preaxial polydactyly. Eur J Hum Genet 2009; 17: 604–610.
Chiang C, Swan RZ, Grachtchouk M et al: Essential role for Sonic hedgehog during hair follicle morphogenesis. Dev Biol 1999; 205: 1–9.
St-Jacques B, Dassule HR, Karavanova I et al: Sonic hedgehog signaling is essential for hair development. Curr Biol 1998; 8: 1058–1068.
Wang LC, Liu ZY, Gambardella L et al: Regular articles: conditional disruption of hedgehog signaling pathway defines its critical role in hair development and regeneration. J Invest Dermatol 2000; 114: 901–908.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR : Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002; 30: 97–101.
Ovcharenko I, Stubbs L, Loots GG : Interpreting mammalian evolution using Fugu genome comparisons. Genomics 2004; 84: 890–895.
Fujioka H, Ariga T, Horiuchi K et al: Molecular analysis of non-syndromic preaxial polydactyly: preaxial polydactyly type-IV and preaxial polydactyly type-I. Clin Genet 2005; 67: 429–433.
Radhakrishna U, Bornholdt D, Scott HS et al: The phenotypic spectrum of GLI3 morphopathies includes autosomal dominant preaxial polydactyly type-IV and postaxial polydactyly type-A/B; No phenotype prediction from the position of GLI3 mutations. Am J Hum Genet 1999; 65: 645–655.
Biesecker LG, Johnston J : Syndromic and non-syndromic GLI3 phenotypes. Clin Genet 2005; 68: 284, author reply 285.
Kroisel PM, Petek E, Wagner K : Phenotype of five patients with Greig syndrome and microdeletion of 7p13. Am J Med Genet 2001; 102: 243–249.
Farooq M, Troelsen JT, Boyd M et al: Preaxial polydactyly/triphalangeal thumb is associated with changed transcription factor-binding affinity in a family with a novel point mutation in the long-range cis-regulatory element ZRS. Eur J Hum Genet 2010; 18: 733–736.
Epstein DJ, McMahon AP, Joyner AL : Regionalization of Sonic hedgehog transcription along the anteroposterior axis of the mouse central nervous system is regulated by Hnf3-dependent and -independent mechanisms. Development 1999; 126: 281–292.
Jeong Y, El-Jaick K, Roessler E, Muenke M, Epstein DJ : A functional screen for sonic hedgehog regulatory elements across a 1 Mb interval identifies long-range ventral forebrain enhancers. Development 2006; 133: 761–772.
Sagai T, Amano T, Tamura M, Mizushina Y, Sumiyama K, Shiroishi T : A cluster of three long-range enhancers directs regional Shh expression in the epithelial linings. Development 2009; 136: 1665–1674.
Amano T, Sagai T, Tanabe H, Mizushina Y, Nakazawa H, Shiroishi T : Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Dev Cell 2009; 16: 47–57.
Galli A, Robay D, Osterwalder M et al: Distinct roles of Hand2 in initiating polarity and posterior Shh expression during the onset of mouse limb bud development. PLoS Genet 2010; 6: e1000901.
Lettice LA, Williamson I, Wiltshire JH et al: Opposing functions of the ETS factor family define Shh spatial expression in limb buds and underlie polydactyly. Dev Cell 2012; 22: 459–467.
Gotea V, Visel A, Westlund JM, Nobrega MA, Pennacchio LA, Ovcharenko I : Homotypic clusters of transcription factor binding sites are a key component of human promoters and enhancers. Genome Res 2010; 20: 565–577.
Zhang Y, Rath N, Hannenhalli S et al: GATA and Nkx factors synergistically regulate tissue-specific gene expression and development in vivo. Development 2007; 134: 189–198.
Durocher D, Charron F, Warren R, Schwartz RJ, Nemer M : The cardiac transcription factors Nkx2-5 and GATA-4 are mutual cofactors. Embo J 1997; 16: 5687–5696.
Shiojima I, Komuro I, Oka T et al: Context-dependent transcriptional cooperation mediated by cardiac transcription factors Csx/Nkx-2.5 and GATA-4. J Biol Chem 1999; 274: 8231–8239.
Molkentin JD, Antos C, Mercer B, Taigen T, Miano JM, Olson EN : Direct activation of a GATA6 cardiac enhancer by Nkx2.5: evidence for a reinforcing regulatory network of Nkx2.5 and GATA transcription factors in the developing heart. Dev Biol 2000; 217: 301–309.
Liu C, Glasser SW, Wan H, Whitsett JA : GATA-6 and thyroid transcription factor-1 directly interact and regulate surfactant protein-C gene expression. J Biol Chem 2002; 277: 4519–4525.
Kozhemyakina E, Ionescu A, Lassar AB : GATA6 is a crucial regulator of Shh in the limb bud. PLoS Genet 2014; 10: e1004072.
Chen Y, Bates DL, Dey R et al: DNA binding by GATA transcription factor suggests mechanisms of DNA looping and long-range gene regulation. Cell Rep 2012; 2: 1197–1206.
Suzuki K, Yamaguchi Y, Villacorte M et al: Embryonic hair follicle fate change by augmented beta-catenin through Shh and Bmp signaling. Development 2009; 136: 367–372.
Ellis T, Smyth I, Riley E et al: Overexpression of Sonic Hedgehog suppresses embryonic hair follicle morphogenesis. Dev Biol 2003; 263: 203–215.
Kurek D, Garinis GA, van Doorninck JH, van der Wees J, Grosveld FG : Transcriptome and phenotypic analysis reveals Gata3-dependent signalling pathways in murine hair follicles. Development 2007; 134: 261–272.
Acknowledgements
We gratefully thank the cell culture platform of the Center of Biology-Pathology, Lille University Hospital (France) for their technical assistance in cell culturing and transfection experiments. This work was supported by funds from Lille University Hospital (France).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Petit, F., Jourdain, AS., Holder-Espinasse, M. et al. The disruption of a novel limb cis-regulatory element of SHH is associated with autosomal dominant preaxial polydactyly-hypertrichosis. Eur J Hum Genet 24, 37–43 (2016). https://doi.org/10.1038/ejhg.2015.53
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.53
This article is cited by
-
Identity-by-descent refines mapping of candidate regions for preaxial polydactyly II /III in a large Chinese pedigree
Hereditas (2018)
-
Why location matters — site-specific factors in rheumatic diseases
Nature Reviews Rheumatology (2017)

{kind=link}
{kind=link}
{kind=link}
{kind=link}