
Abstract
Vascular Ehlers–Danlos syndrome (vEDS) is a rare and severe autosomal dominant disorder caused by variants at the COL3A1 gene. Clinical characteristics and course of disease of 215 molecularly proven patients (146 index cases and 69 relatives) were analysed. We found 126 distincts variants that were divided into five groups: (1) Glycine substitutions (n=71), (2) splice-site and in-frame insertions–deletions (n=36), (3) variants leading to haplo-insufficiency (n=7), (4) non-glycine missense variants within the triple helix (n=4 variants), and (5) non-glycine missense variants or in-frame insertions–deletions, in the N- or C-terminal part of the protein (n=8). Overall, our cohort confirmed the severity of the disease with a median age at first complication of 29 years (IQR 22–39), the most frequent being arterial (48%) and digestive (24%) ruptures. Groups 2 and 1 were significantly more severe than groups 3–5, with extreme median ages at first major complication of 23–47 years. Patients of groups 3–5 had a less typical phenotype and remarkably absence of digestive events. The distribution of glycine-replacing amino acids was strongly biased towards more destabilizing residues of the collagen assembly. Thus the natural course of vEDS and the clinical phenotype of patients are influenced by the type of COL3A1 variant. This study also confirms that patients with variants located in the C- and N-termini or leading to haplo-insufficiency have milder course of the disease and less prevalent diagnostic criteria. These findings may help refine diagnostic strategy, genetic counselling and clinical care.
Similar content being viewed by others

Introduction
Vascular Ehlers–Danlos syndrome (vEDS, OMIM #130050) is a rare inherited autosomal dominant disorder with an estimated prevalence of 1/150 000. Patients are exposed to life-threatening complications, typically spontaneous arterial dissections and ruptures, bowel perforations and uterine ruptures.1, 2 Clinical events usually occur from early adulthood onwards with unpredictable relapse intervals. Each complication is at high risk of death, and consequently, life expectancy is reduced, estimated in the early 2000s to a median of 48 years.2
Major and minor clinical diagnostic criteria of the Villefranche classification illustrate the variety of physical signs that may constitute the clinical phenotype and explain the difficulty of its diagnosis.3 Formal diagnosis of vEDS requires a positive genetic test, that is, the demonstration of a pathogenic variant within the COL3A1 gene encoding the pro-alpha 1 chain of type III procollagen. More than 570 COL3A1 genetic variants (including the 126 described in the present article) are registered in the Ehlers–Danlos Syndrome Variant Database, a large majority of them being private.4 Most of the variants are heterozygous missense substitutions affecting one of the glycine residues of the [Gly-X-Y]343 repeat within the triple helical region of type III collagen, acting as dominant-negative variants.2 vEDS might also be caused by heterozygous splice-site variants of exons encoding a triple helix sequence, the vast majority leading to an in-frame exon skipping and generation of a shorted translated product also altering the stable assembly of type III procollagen homotrimers.5 The rare frameshift, non-sense or large deletion variants result in a reduction of mature procollagen and haplo-insufficiency.6 Ultimately, both the alteration of type III collagen synthesis and assembly result in decreased resistance to mechanical stress of hollow organs, especially arteries and bowel.7, 8
Despite clinical and genetic heterogeneity, only variants leading to haplo-insufficiency have been discussed to result in a milder phenotype.5, 6, 9 In order to investigate the possibility of other associations between the nature and location of COL3A1 variants and the phenotypic expression of the disease, the clinical characteristics and major complications in 215 patients with molecularly proven vEDS were analyzed.
Patients and methods
Study population
Patients with vEDS referred between 2001 and 2014 to the French National Reference Centre for Rare Vascular Diseases and to the corresponding network of referral centres for either clinical care or genetic diagnosis were included in a retrospective cohort study. We have previously described the oral phenotype of 17 of them.10 All patients had a standardized assessment of personal medical history and a physical examination by a senior physician. During genetic counselling, first-degree relatives were proposed to be examined and genetically screened.
The patients' clinical characteristics were staged as major or minor diagnostic criteria according to the Villefranche nosology.3 Vascular, digestive and/or obstetrical events were assessed until last known follow-up visit in our department or in referring centres. Arterial events were defined by symptomatic dissections, aneurysms or spontaneous arterial ruptures. Digestive events were defined by spontaneous bowel perforations or ruptures requiring surgery. All postoperative or likely surgery-related digestive events were excluded from analysis. Obstetrical events were defined by uterine ruptures, arterial ruptures or dissections occurring during pregnancy, delivery or in the 1-month postpartum period.
Genetic testing was performed in compliance with French legislation on genetics diagnostics tests (French bioethics law no. 2004–800), and written informed consent was obtained for all patients. Decision of genetic testing was made after a multidisciplinary outpatient visit, associating a geneticist, a specialist in vascular medicine and a genetic counsellor. Psychological support was organized for each patient before and throughout the genetic testing process when necessary.
Genetic analysis
COL3A1 gene was screened for all patients either by direct sequencing from genomic DNA (gDNA) and Multiplex Ligation Probe Amplification or by sequencing from complementary DNA (cDNA) (for details of the genetic analysis methods, see Supplementary Method S1, Supplementary Tables S1 and S2). When a variant was detected, independent direct sequencing or arrayCGH was carried out as a control, when possible. Variants at the nucleotide and protein levels are described according to the nomenclature recommended by the Human Genome Variation Society (www.hgvs.org/mutnomen), based on the COL3A1 human cDNA and gene sequences (GenBank NM_000090.3 and NG_007404.1), which number the COL3A1 gene exons from 1 to 51. Variants were registered in the Ehlers–Danlos Syndrome Variant Database (https://eds.gene.le.ac.uk/variants.php?action=search_unique&select_db=COL3A1).
Statistical analysis
Patients’ characteristics are reported for the whole cohort and according to the ascertainment status (index case or relative) and the type of variant. Time-independent qualitative variables were compared using chi-square test or Fisher’s exact test (when count was insufficient in the contingency table). The comparison of the incidence onset of the major complications was performed using Kaplan–Meier survival curves with Log-rank test. In respect of the comparison between the types of variant, Log-rank tests were stratified for the ascertainment status. Quantitative variables were reported using non-parametric descriptive statistics (median, interquartile range (IQR)) and compared using the Wilcoxon test. A sensitivity analysis was performed by replicating all comparisons in the index patients group only. A P-value <0.05 was considered significant. The statistical analysis was performed with the R software (version 3.1.1).11
Results
Clinical diagnostic criteria of index cases and first-degree relatives
A total of 215 patients (146 index cases, 69 first-degree relatives) were studied (Table 1). Almost all patients were of European origin and subjects were predominantly women (62%). The age of occurrence of the first major event did not differ significantly between men and women (Supplementary Figure S1). The median age at diagnosis was 33 (IQR 24–43) years for index cases and 38 (IQR 19–49) for relatives. As a consequence of recruitment bias, almost all index cases (90%) had experienced at least one complication at diagnosis compared with only 58% of relatives (P<0.001). They had also more frequent major diagnostic criteria (thin and translucent skin, extensive bruising and characteristic facial appearance) (Table 1). The median prevalence of each minor diagnostic criteria was similar in both index cases and relatives, except for acrogeria and the occurrence of carotid cavernous fistula (Supplementary Table S3).
Survival analysis showed significant differences in the age at which the first major event occurred in index cases and relatives (Figure 1). At 28 years, half of index cases had experienced a first major complication, versus 25% of relatives only. At 43 years, half of relatives had experienced at least one major complication versus 85% of index cases.

Age of onset of the first major complication in 215 vEDS patients.
First and second major events during the follow-up
In symptomatic patients, first major events were predominantly arterial (63%), followed by digestive complications (26%) (Table 2a). In women, the disease was revealed by a first obstetrical event in 12% of the cases. Arterial complications were predominantly local dissections and aneurysms at the iliac, renal and carotid arteries (80% with multiple locations), whereas digestive complications were spontaneous colonic perforation in 85% of the cases. We also observed that the second major event was influenced by the type of first event (Table 2b). It was more frequently digestive for patients with a first gastrointestinal event than for patients with a first arterial event (39 versus 7%, respectively; P<0.0001).
COL3A1 variants distribution and classification

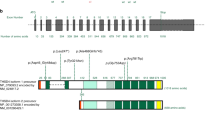
In the n=146 index cases, 126 distinct variants were identified by Sanger sequencing on gDNA (Figure 2). Fifty of them were confirmed by cDNA sequencing (Supplementary Table S4). Variants were spread along the entire COL3A1 gene, 90% (n=114 variants) of them were located between exons 6 and 47 encoding the triple helix domain of type III procollagen. More than half of the 126 variants (n=68) were not previously reported, including the complete gene deletion. The majority of them were glycine substitutions (Group 1; 71 distinct variants). The second most frequent variants were splice-site variants, which were predominantly (83%) located at the 5′ donor site, or insertions/deletions resulting in a large in-frame anomaly located in the triple helix (Group 2; 36 distinct variants). We classified the other variants in three other groups: those resulting in haplo-insufficiency (group 3; 7 distinct variants), non-glycine missense variants located in the triple helix (group 4; n=4), and non-glycine missense variants located in the C- or N-terminal regions of the protein (group 5; n=8).

COL3A1 gene variants detected in 146 unrelated index cases with vEDS. Boxes represent exons of the COL3A1 gene (NG_007404.1) with colour code representing protein domains (yellow: N- and C-terminal domains, green: transition domains, blue: collagen helical domain). Glycine substitutions are presented on the left in a shortened, nonofficial nomenclature. The complete nomenclature of each variant can be found in the Supplementary Table S4, in which variants detected on the genomic but not on the cDNA sequence are indicated with parentheses, for example, p.(Gly324Ser). All of them have been reported in the Ehlers–Danlos Syndrome Variant Database (see Methods, Genetic analysis). Substitutions for other amino acids, splice-site variants and insertions/deletions are shown on the right. indicates the variants found more than once in the index cases cohort; †indicates previously undescribed variants.
Most of the point substitutions were private and only 12 of them were common to several unrelated families (Figure 2). Among them, the splice-site variant at intron 23, c.1662+1G>A, showed the highest level of recurrence in seven independent families (Supplementary Table S4a). Two splice-site variants, located at intron 48 and 49 5′ donor sites, c.3283+1G>C (patient AN_002976) and c.4011+1G>A (patient AN_002972) led to an out of frame exon skipping, the occurrence of a premature termination codon and finally resulted in haplo-insufficiency. Details of the variants and their locations are presented in Figure 2, except for the complete deletion of the COL3A1 gene found in one index case (see Supplementary Table S4 for detailed genotypic and phenotypic analysis of patients of each group).
Genotype–phenotype associations
According to the Villefranche nosology, the number of major criteria was consistent among groups 1–4 but significantly lower in group 5 (Table 3). However, the number of minor criteria differed according to the groups of patients (Supplementary Table S5): compared with group 1 (glycine substitutions), group 2 (splice-site variants) had more minor criteria whereas groups 3–5 had fewer (P<0.01 for each comparison). However, when evaluated independently, the median prevalence of each minor diagnostic criteria was similar in all groups except for acrogeria, which was more prevalent in groups 1 and 2, when compared with groups 3–5 (P<0.01).
Similarly, the age at diagnosis was lower in the groups 1–2 as compared with the three other groups (Table 3, global P-value <0.001): group 2 had a low median age at diagnosis (25 years (18–33)), followed by group 1 with a delay of 10 years (34 years (27–42)). The groups 3–5 had a similar age at diagnosis, delayed by 12 years from group 1 (46 (38–52) years, 45 (44–48), and 45 (38–49), respectively). The same distribution was observed for the age at first major event. At the age of 40 years, proportion of event-free survival was 10% for patients of group 2, 24% in group 1; 70% in group 3; 80% in group 4 and 55% in group 5 (P<0.001, Figure 3). Interestingly, none of the patients of groups 3–5 had digestive complications, versus 36 and 34% in groups 1 and 2, respectively (global P<0.001, Table 3).

Age of onset of the first major complication as a function of type of COL3A1 variant in 215 patients with vEDS. Abbreviations: Splice/del/ins/dup, insertion, deletion, duplication or splice-site variant in the triple helix; Gly, Glycine; C/N domain, C-terminal or N-terminal domain of the protein; TH, triple helix.
We also analysed the nature of the amino acids substituting the glycine residues, as a bias for several types of residues was previously observed for α(I) and α(III) collagen chains in relation with the extent of destabilization of the triple helix.12 The analysis of n=79 index cases with Gly missense variants showed that the most destabilizing Asp, Val and Glu amino acids were over-represented (n=29, 36%; n=18, 23%; n=9, 11%; respectively), whereas Ser and Ala residues were under-represented (n=11, 15%; n=1, 1%; respectively, global P<0.0001) (Figure 4, Supplementary Table S6). This bias towards the more destabilizing residues was tested as to whether they were associated with a more severe phenotype in all patients. No marked significant difference was observed between the three subgroups (Asp/Val/Glu, Arg, Ser–Ala), except for the characteristic facial appearance (P=0.003) and extensive bruising (P=0.012) that could possibly be due to the relatively low number of subjects per subgroup (Supplementary Table S7).

Distribution of observed and expected AA residues substituting a Glycine residue. Expected substitutions are calculated from the corrected expected numbers as previously reported.12 Yates’ chi-squared, P<0.001.
Discussion
We herein describe the largest series of European patients with molecularly proven vEDS systematically screened in the French referral Centre for this rare inherited disease. Our findings confirm the severity of the disease, positive individuals having experienced a first major vascular, digestive or obstetrical complication at the median age of 29 years. We show that gender does not influence the prognosis. More importantly, we identified subgroups of patients who have a later-onset of the disease and a better prognosis, and especially no digestive events. The five subgroups of variants have also differences in some major and minor criteria, acrogeria, characteristic facial appearance, thin translucent skin and extensive bruising being hallmarks of a severe disease. Finally, we show that for missense variants affecting a glycine residue, the distribution of mutated amino acids is strongly biased towards residues having the stronger destabilizing effect on collagen assembly.
The occurrence of clinical events, their frequency and distribution according to age were comparable to the available cohorts published in 2000 and 2014.2, 9 Overall, 17% of our vEDS patients had a first complication by the age of 20 years, and 71% had at least one major complication by the age of 40 years. We did not observe a significant gender effect, contrary to the recent survival analysis made by Byers’s group.13 However, our cohort differs from the US cohort2, 13 in several aspects because of different recruitment and clinical monitoring modalities: index patients and their relatives described here were approximately 5 years older at the time of diagnosis, and number of symptom-free index cases at referral was only 11% in this cohort versus 39% in the US cohort. This important difference has multiple explanations: patients older in our cohort, possible referral bias for more severe subjects, increasing awareness of the disease, and improved imaging modalities and accessibility of genetic testing. We did not analyse intra-familial phenotypic heterogeneity or penetrance in this study, because we chose to limit the number of patients per family to three in order to avoid familial phenotypic bias. However, prospective systematic evaluation of individuals with the same variant currently performed in our centre may give further insight in the near future.
Interestingly, we found that some groups of patients are more prone to specific complications. First, all patients with digestive ruptures belonged either to group 1 (n=44, 36% of group 1 patients) or to group 2 (n=19, 34%). Patients belonging to the three other groups of variants had no digestive event at the median age of 45 years, whereas all colonic ruptures in groups 1 and 2 were observed in early adulthood (median 23 years, range 19–34). It is noteworthy that we observed no familial aggregation for digestive events. Second, as recently presented by Shalhub et al,9 aortic complications (aneurysms, dissections and ruptures) appear to be more prevalent in patients with haplo-insufficiency (4/8, 50% of index cases), when compared with glycine substitution (19/79, 19%) or splice-site variant (6/45, 13%) patients (global P=0.02).
Genetic screening of the COL3A1 gene showed a majority of missense variants involving a glycine residue (54% of index cases), followed by splice-site variants (31%), in agreement with previous reports and with the Ehlers–Danlos Syndrome Variant Database (https://eds.gene.le.ac.uk/home.php?select_db=COL3A1). The affected glycine residues were spread from exons 6 to 47, the region encoding chains of the triple helix domain of type III procollagen. As expected, almost all of these variants were private, only 5 out of the 71 glycine substitutions being recurrent. Whether the location of the mutated glycine residue could be associated to the phenotype has been the focus of previous studies. By the analysis of a limited set of 14 different glycine substitutions, Pope et al1 found an increasing abnormal phenotype when the variant shifted from exon 36 to exon 49. We could not confirm this tendency in this larger cohort of affected individuals (data not shown), consistently with the findings observed in the even greater US cohort.2, 13
We also found a strong distribution bias of the glycine replacements, consistently with previous reports suggesting that for the COL3A1 gene the most destabilizing variants are over-represented in comparison to less destabilizing residues.12 Indeed, in our series 73% (58/79) of the mutated residues in index cases were either Val, Glu or Asp, much more frequent than Ala and Ser (18/79, 23%), proportions that diverge significantly from that expected of the possible outcomes of a single base pair substitution in a glycine codon (P<0.001 for the two subgroups). This selection bias that was also recently observed by Pepin et al13 cannot be explained by the transition/transversion rate bias in the human coding sequences and is positively correlated to the triple helix destabilizing effect of the corresponding variants.14 The collagen triple helical structure is indeed disrupted more profoundly when Gly is replaced by a large charged amino acid such as Asp, than by a smaller residue such as Ala. A recent in vitro study tested the effect of two types of missense variants (Gly-to-Val and Gly-to-Ala) on the type III collagen folding using a bacterial system for production of homotrimeric model polypeptides.15 The effect of the Gly-to-Val variant was stronger compared with Gly-to-Ala, suggesting that some Ala variants might not be severe enough to cause vEDS.
The distribution of splice-site variants was characterized by an over-representation of variants at the 5′ donor site (30 out of the 36 distinct variants) as already observed.13, 16 Eighteen out of the 36 distinct variants were already shown to lead to exon skipping or to cryptic splice-site activation and to decreased secretion of procollagen trimers.16, 17 Even though there was no variant hotspot in our cohort, a few recurrent substitutions were present, especially the splice-site variant at intron 23, c.1662+1G>A, which is also the most frequent splice-site variant recorded in the Ehlers–Danlos Syndrome Variant Database. In our series, patients with splice-site variants had an earlier age at diagnosis than patients with glycine substitutions (25 versus 34 years, P=0.0002) and a comparable high prevalence of clinical events, of major and of minor diagnostic criteria. Almost all of these splice variants are in phase and lead to a product deleted of 54–108 residues that can act in a dominant-negative manner, strongly destabilizing the collagen assembly. To our best knowledge, whether this deleterious effect is more important than the one caused by glycine substitutions has not been investigated in biochemical studies.
One of the main findings of our study was the identification of yet unreported missense variants (groups 4 and 5) that were associated with mild clinical features of vEDS and arterial fragility. In addition to being evidenced in patients with suggestive features of the pathology, we also found arguments for their possible pathogenicity, which are given in the Supplementary Materials (Supplementary Results and Methods, Supplementary Tables S4c–e). Importantly, variants at the N- and C-terminal parts of the protein and the missense variants in the triple helix allowed identifying a subgroup of patients representing 10% of our cohort. These patients displayed a milder clinical phenotype with sometimes only one major and/or one minor clinical criterion. However, despite this mild clinical course, the absence of digestive complications and the presence of some of these variants in population databases at very low frequency, our findings suggest that these patients are at higher risk of arterial events and should be considered and followed as such. Heterogeneity in phenotypic expression in vEDS has been suggested previously, ranging from variable clinical phenotypes for non-glycine missense variants located in the C-terminal domain to overlapping with hypermobile EDS for non-glycine variants located in the triple helix domain of type III collagen.1, 18 As for patients with Marfan syndrome that remained undiagnosed when staged by the Ghent nosology prior to its latest revision,19 it may be necessary to broaden indications for genetic testing in vEDS. For example, patients with only one major criterion and a familial history of sudden death, or patients with a major vascular event before the age of 50 years even in the absence of evocative clinical signs (as evidenced by the absence of clinical signs of the disorder in the relatives versus the index cases of our cohort (Table 1)),20 may be discussed as candidates for COL3A1 genetic screening, once common underlying causes of cardiovascular disease have been ruled out. As we specifically showed in group 3, the absence of digestive events associated with a high prevalence of aortic complications could mimic familial thoracic aortic aneurysm and dissection (FTAAD, OMIM #%607086 for general informations). It is important to suspect vEDS also in these patients as therapeutic decisions are different from the ones for other inherited thoracic aortic aneurysms and dissections. Recent progress in sequencing technology should facilitate this wider genetic screening, even though some variants will require biochemical studies to establish their causal role on type III collagen alteration of assembly and/or production.
Conclusion
This study confirms in a large European cohort fully characterized in a dedicated diagnostic network that vEDS is a severe early-onset disease. In addition to the previously described subgroup of patients with variants leading to haplo-insufficiency, it also identifies subgroups of patients bearing non-glycine missense and/or genetic variations at the C- and N-termini of type III procollagens, which are characterized by a later-onset and a milder phenotype, with no or very rare digestive complications. These findings may affect diagnostic strategy, genetic counselling and clinical follow-up of patients with vascular Ehlers–Danlos syndrome.
References
Pope FM, Narcisi P, Nicholls AC, Germaine D, Pals G, Richards AJ : COL3A1 mutations cause variable clinical phenotypes including acrogeria and vascular rupture. Br J Dermatol 1996; 135: 163–181.
Pepin M, Schwarze U, Superti-Furga A, Byers PH : Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med 2000; 342: 673–680.
Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ : Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet 1998; 77: 31–37.
Dalgleish R : The Human Collagen Mutation Database 1998. Nucleic Acids Res 1998; 26: 253–255.
Leistritz DF, Pepin MG, Schwarze U, Byers PH : COL3A1 haploinsufficiency results in a variety of Ehlers-Danlos syndrome type IV with delayed onset of complications and longer life expectancy. Genet Med 2011; 13: 717–722.
Schwarze U, Schievink WI, Petty E et al: Haploinsufficiency for one COL3A1 allele of type III procollagen results in a phenotype similar to the vascular form of Ehlers-Danlos syndrome, Ehlers-Danlos syndrome type IV. Am J Hum Genet 2001; 69: 989–1001.
Prockop DJ, Kivirikko KI : Heritable diseases of collagen. N Engl J Med 1984; 311: 376–386.
Bornstein P, Sage H : Structurally distinct collagen types. Annu Rev Biochem 1980; 49: 957–1003.
Shalhub S, Black JH 3rd, Cecchi AC et al: Molecular diagnosis in vascular Ehlers-Danlos syndrome predicts pattern of arterial involvement and outcomes. J Vasc Surg 2014; 60: 160–169.
Ferre FC, Frank M, Gogly B et al: Oral phenotype and scoring of vascular Ehlers-Danlos syndrome: a case-control study. BMJ Open 2012; 2: e000705.
CoreTeam A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, 2013.
Persikov AV, Pillitteri RJ, Amin P, Schwarze U, Byers PH, Brodsky B : Stability related bias in residues replacing glycines within the collagen triple helix (Gly-Xaa-Yaa) in inherited connective tissue disorders. Hum Mutat 2004; 24: 330–337.
Pepin MG, Schwarze U, Rice KM, Liu M, Leistritz D, Byers PH : Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet Med 2014; 16: 881–888.
Beck K, Chan VC, Shenoy N, Kirkpatrick A, Ramshaw JA, Brodsky B : Destabilization of osteogenesis imperfecta collagen-like model peptides correlates with the identity of the residue replacing glycine. Proc Natl Acad Sci USA 2000; 97: 4273–4278.
Mizuno K, Boudko S, Engel J, Bachinger HP : Vascular Ehlers-Danlos syndrome mutations in type III collagen differently stall the triple helical folding. J Biol Chem 2013; 288: 19166–19176.
Schwarze U, Goldstein JA, Byers PH : Splicing defects in the COL3A1 gene: marked preference for 5′ (donor) spice-site mutations in patients with exon-skipping mutations and Ehlers-Danlos syndrome type IV. Am J Hum Genet 1997; 61: 1276–1286.
Giunta C, Steinmann B : Characterization of 11 new mutations in COL3A1 of individuals with Ehlers-Danlos syndrome type IV: preliminary comparison of RNase cleavage, EMC and DHPLC assays. Hum Mutat 2000; 16: 176–177.
Narcisi P, Richards AJ, Ferguson SD, Pope FM : A family with Ehlers-Danlos syndrome type III/articular hypermobility syndrome has a glycine 637 to serine substitution in type III collagen. Hum Mol Genet 1994; 3: 1617–1620.
De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE : Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet 1996; 62: 417–426.
Faivre L, Collod-Beroud G, Callewaert B et al: Pathogenic FBN1 mutations in 146 adults not meeting clinical diagnostic criteria for Marfan syndrome: further delineation of type 1 fibrillinopathies and focus on patients with an isolated major criterion. Am J Med Genet A 2009; 149A: 854–860.
Acknowledgements
In memory of Dr Jerome Perdu who initiated the Centre for Rare Vascular Diseases at the Hôpital Européen Georges Pompidou in Paris, France and was involved in the care of several patients and families reported here. We thank all patients and families and the Association Française des Syndromes d’Ehlers–Danlos (AFSED) for their confidence and continuing support. We acknowledge the contributions of clinicians of the 'Centres de Compétence' of the French network of vEDS patients. We also thank the technicians of the genetics laboratory (Magali Boussion, Valérie Boccio, Annabelle Venisse, Nelly Lepottier, Valérie Nau, Isabelle Roncelin) for their contribution to the COL3A1 genetic analysis. This study was supported by the Assistance Publique des Hôpitaux de Paris, the INSERM, the French Ministry of Health through the program 'Maladies Rares 2006' and the Soutien aux Technologies Innovantes et Coûteuses (STIC 2006), the Agence Nationale pour la Recherche (10-BLAN-1129 GDPM2) and by the European Community’s 7th Framework Program (HEALTH-F2-2007-Fighting Aneurysm Disease program).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Frank, M., Albuisson, J., Ranque, B. et al. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers–Danlos syndrome. Eur J Hum Genet 23, 1657–1664 (2015). https://doi.org/10.1038/ejhg.2015.32
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.32
This article is cited by
-
Diagnosis and treatment of cardiovascular disease in patients with heritable connective tissue disorders or heritable thoracic aortic diseases
Cardiovascular Intervention and Therapeutics (2024)
-
Diagnosis and management of vascular Ehlers-Danlos syndrome: Experience of the UK national diagnostic service, Sheffield
European Journal of Human Genetics (2023)
-
Vascular Ehlers-Danlos syndrome with a Novel missense COL3A1 gene mutation present with bilateral spontaneous carotid-cavernous fistula: a case report
Acta Neurochirurgica (2023)
-
Efficient C•G-to-G•C base editors developed using CRISPRi screens, target-library analysis, and machine learning
Nature Biotechnology (2021)
-
Genetische Analysen bei hereditären Bindegewebserkrankungen
Journal für Mineralstoffwechsel & Muskuloskelettale Erkrankungen (2021)

{kind=link}