
Abstract
Chronic intestinal pseudo-obstruction (CIPO) syndromes are heterogeneous gastrointestinal disorders, caused by either neuropathy or myopathy, resulting in compromised peristalsis and intestinal obstruction. CIPO can have a profound impact on quality of life, leading the most severely affected individuals to life-long parenteral nutrition and urinary catheterization. To search for disease causing gene(s), we performed the whole exome sequencing (WES) in both eight sporadic and two familial cases, followed by targeted sequencing in additional CIPO patients. After identifying a heterozygous missense variant in the ACTG2 gene in one of 10 patients undergone WES, targeted Sanger sequencing of this gene allowed to detect heterozygous missense variants in 9 of 23 further patients with either megacystis-microcolon-intestinal hypoperistalsis syndrome or intestinal pseudo-obstruction. Variants thus identified, one of which still unreported, affect highly conserved regions of the ACTG2 gene that encodes a protein crucial for correct enteric muscle contraction. These findings provided evidence for a correlation between the clinical phenotype and genotype at the ACTG2 locus, a first step to improve the diagnosis and prognosis of these severe conditions.
Similar content being viewed by others

Introduction
Gastrointestinal motility disorders range from common and generally benign to rare and potentially life-threatening diseases.1 Neuropathy, myopathy or defective interstitial cells of Cajal (ICC) can underlie these conditions, resulting in compromised peristalsis leading to chronic intestinal pseudo-obstruction (CIPO) syndromes.1, 2 Intestinal neuronal dysplasia type B (INDB), characterized by aspecific derangements of the enteric nervous system (ENS) with submucosal hyperganglionosis, is a controversial entity, which may also be responsible for CIPO.1
Although most CIPO patients show a sporadic occurrence, rare familial transmissions support a genetic etiology.2 Candidate genes have already been tested in patients,3, 4 however, only a small proportion of the genetic heterogeneity of CIPO could be resolved.5, 6 Variants of the ACTG2 gene, encoding gamma 2 enteric actin, a protein crucial for correct enteric muscle contraction, have been found in CIPO patients affected with congenital or late-onset visceral myopathy or, alternatively, megacystis-microcolon-intestinal hypoperistalsis syndrome (MMIHS).7, 8, 9, 10, 11, 12 Here we report a study, carried out by whole exome sequencing (WES) and targeted Sanger sequencing in a total of 33 CIPO patients, which has allowed to define part of the phenotypic spectrum associated with ACTG2 defects.
Subjects and methods
For details about subjects under analysis and methodologies, see the Supplementary File. Genetic data were submitted to ClinVar (www.ncbi.nlm.nih.gov/clinvar/) and the following accession numbers were obtained for the ACTG2 variants detected: SCV000256210 (p.R178H), SCV000256211 (p.R178L), SCV000256212 (p.R178C), SCV000256213 (p.R257H), SCV000256214 (p.R257C), SCV000256215 (p.R38H) and SCV000256216 (p.R148S).
Results
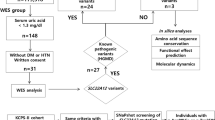
We have conducted a study aimed at identifying the genetic cause(s) of CIPO in 30 sporadic patients and three families, some of whom had previously been reported.3, 13, 14 Patients were affected with intestinal dysmotility and most of them already characterized from a clinical and histopathological point of view. With the exception of three sporadic patients affected with late onset visceral myopathy, referred to us by adult gastroenterologists and already tested for candidate genes,13 all the other patients had had a pediatric onset of the disease.
WES was applied to a first set of CIPO patients, including eight sporadic cases and the index cases of two families. Of these 10 unrelated patients, one presented with intestinal pseudo-obstruction and megacystis and nine with CIPO, including three diagnosed with INDB. A heterozygous missense variant of the ACTG2 gene was identified in the former patient. Examination of the exome data set of the remaining 9 samples showed that all the coding exons and splice sites of ACTG2 were properly covered and did not harbor any variant.
Subsequently, by targeted Sanger sequencing, we detected heterozygous missense variants in 9 cases with either MMIHS or CIPO. A summary of all the heterozygous ACTG2 variants found in our cohort together with clinical features is reported in Table 1. Since the parents of five probands did not carry any variant, we concluded that these variants occurred de novo, at least in those cases. We did identify one new missense variant not yet reported in the literature, namely the nucleotide substitution c.113G>A (p.(Arg38His)), in patient S24 (Table 1). According to online tools predicting functional effect of human missense variants, such as Sift (http://sift.jcvi.org/) and Polyphen (http://genetics.bwh.harvard.edu/pph2/), this variant is a putative damaging variant at a highly conserved region in the ACTG2 gene. Moreover, it is reported neither in the Exome Sequence Variant database (http://evs.gs.washington.edu/EVS/), the Exome Aggregation Consortium (ExAC) database (Cambridge, MA, USA—http://exac.broadinstitute.org—October 2015 accessed) nor in the Single Nucleotide Polymorphism database (dbSNP 142—http://www.ncbi.nlm.nih.gov/snp/). Interestingly, the above patient, S24, and two other patients, S8 and S9, with the ACTG2 c.770G>A (p.(Arg257His)) variant (Table 1), had a histological evaluation that fulfilled the diagnostic criteria for INDB.1
Histopathological reassessment (see Supplementary File for details) was performed on colectomy specimens from the S9 patient. In addition to severe atrophy of both layers of the muscularis propria in the proximal colon, an almost complete absence of connective fibers network was also revealed. Microtubule-associated protein-2 (MAP-2) ruled out hyperganglionosis and confirmed the presence of a normal number of ganglia though, in keeping with the original report of INDB, at least 20% of them contained more than eight cells (Figure 1).

Representive histopathological pictures of the colon obtained from patient S9 (see Table 1). (a) Note a clearly detectable change in the thickness and structural organization of the muscularis propria of the distal (right) and proximal (left) colon; notably the muscle thickness in the distal colon exceeds that of the proximal colon. Hematoxylin and eosin staining. Original magnification 50x for both right and left pictures. The lower panel shows a significant reduction of the smooth muscle actin (SMA) immunolabeling in the distal colon (right); SMA immunolabeling was even absent in the most affected proximal colon (left). Original magnification was 50 × for both right and left pictures. (b) MAP-2 immunohistochemical staining (400 ×) shows the presence of giant ganglia. A full color version of this figure is available at the European Journal of Human Genetics journal online.
On the other hand, the remaining 20 sporadic CIPO patients, as well as three probands, each from one of the three families with CIPO recurrence (Supplementary File), resulted negative for ACTG2 variants (Table 2).
Since a homozygous nonsense variant in the MYH11 gene, coding for the smooth muscle myosin heavy chain, has been identified in one MMIHS patient from consanguineous parents,15 we searched for variants of this gene in our 3 familial cases without finding any variant (not shown).
Discussion
CIPO includes a heterogeneous group of disorders that are still very challenging in terms of diagnostic assessment, possible therapeutic interventions and genetic counseling to families. The diagnosis of CIPO is often difficult because of (i) it still relies on clinical experience rather than on biomarkers of disease, (ii) the clinical presentation (intestinal sub-occlusive crisis mimicking a mechanical sub-occlusion) and complexity of the clinical picture (ie, the presence of co-morbidities such as eg, the urinary bladder abnormalities or the syndromic forms) and, finally, (iii) the wide heterogeneity of mechanisms leading to CIPO and related clinical manifestations.2 In addition, the extremely low incidence of CIPO further adds uncertainty to the clinical spectrum of these patients. In this light, we aimed at identifying genetic cause(s) and took advantage of the availability of a pediatric patient set in our Institute, which is remarkably large given the rarity of CIPO.
Results achieved during this study confirm the importance of the contractile apparatus of the smooth muscle in gastrointestinal motility disorders. Nevertheless, our findings are remarkable for several reasons.
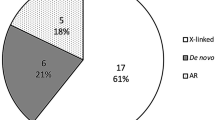
First, given 10 probands carrying ACTG2 variants in comparison with 23 probands without ACTG2 variants, we can conclude that ACTG2 variants underlie a significant proportion of CIPO phenotypes. While a recessive inheritance had been suggested for some MMIHS familial clusters,16 our patients appear to follow a putative dominant pattern of inheritance with de novo fully penetrant variants. However, we can only speculate about underlying pathogenic mechanisms. Given the majority of causative de novo ACTG2 variants detected in the present patients are precisely clustered in only four codons, interestingly coding for arginine residues (Arg38, Arg148, Arg178 and Arg257), either a gain-of-function or a dominant negative pathogenic mechanisms can be invoked for the resulting disorders.
Indeed, the reported presence of abnormal intracellular inclusion bodies and abnormal muscularis propria architecture with reduction of the normal protein in patients with a p.(Arg148Ser)) ACTG2 variant7, 8 can consistently rule out different pathogenic hypotheses.
Furthermore, an altered cellular ACTG2 staining pattern in smooth muscle cells of the muscularis propria, with abnormal aggregates or clumps of stain in the cytoplasm resulting in a granular appearance, possibly due to an accumulation of unpolymerized, non-functional ACTG2 fibers in the inner layer, was reported also in a p.(Arg257His) ACTG2 variant carrier.12 Consistently, the histological reassessment of our patient S9, carrying the same p.(Arg257His) variant, showed abnormal muscularis propria architecture and abnormal smooth muscular actin expression. On the other hand, no anomaly of muscle layers was reported in a patient affected by visceral myopathy and carrying a p.(Gly269Glu) ACTG2 variant.11 In addition, cell experimental studies with ACTG2 variant constructs revealed reduced incorporation of the variant protein into actin filaments7 and confocal images allowed to assess a poor association of ACTG2 mutant fibers with actin filaments,9 thus suggesting again a dominant negative effect of the ACTG2 variants.
Second, genotype–phenotype correlation can be attempted. With only one exception, all the patients reported here with ACTG2 variants had neonatal/infantile onset with gastrointestinal motility disorder, intractable constipation, urinary catheterization and dependence on total parenteral nutrition. Differently from Wrangler et al.,10 who classified as MMIHS also pseudo-obstruction patients with no megacystis and microcolon, here we refer to cases where the presence of microcolon could not be confirmed as CIPO+megacystis (CIPO+M) rather than as MMIHS. This has allowed us to correlate the clinical phenotype with the genotype at the ACTG2 locus, finding that variants affecting codon Arg178, namely c.533G>A (p.(Arg178His)), c.533G>T (p.(Arg178Leu)) and c.532C>T (p.(Arg178Cys)), appear to be related to the most severe form of CIPO, namely MMIHS. Table 1 shows that missense substitutions of codon Arg257, namely c.769C>T (p.(Arg257Cys)) and c.770G>A p.(Arg257His)), are associated with patients showing CIPO with megacystis, a condition less severe than MMIHS though still life-threatening and with a poor outcome. Given the disparities in the clinical classification of CIPO between different patient sets, the genotype–phenotype correlation we have deduced here mainly applies to our dataset. Noteworthy, observations confirming our hypothesis have been reported for the highly homologous ACTA2 gene, encoding the predominant α2-actin isoform in the smooth muscle of the vascular wall. p.(R258H) and p.(R258C) in ACTA2 have been implicated as disease causing variants in families with thoracic aortic aneurysm and dissections.17 On the other hand, the p.(R179H) variant in ACTA2 causes severe multisystem smooth muscle cell dysfunction with early-onset vascular disease similar to moyamoya disease,18 and has also been reported in a patient with prune belly sequence and first trimester-onset fetal megacystis.19 Interestingly, variants of Arg179 in ACTA2 cause more severe phenotypes than variants of Arg258,17 thus confirming similar effects of substitutions at paralogous nucleotide positions of ACTA2 and ACTG2 genes and, therefore, the genotype–phenotype correlation we propose for variants at residues Arg257 and Arg178 of the ACTG2 gene.
The cases of variants of codons Arg38 and Arg148 need to be also considered. The c.113G>A (p.(Arg38His)) variant of our set apparently displays a milder effect, with the affected patient showing only CIPO and no further complications. Consistent with previous observations of adult patients carrying variants affecting codon Arg148,7, 8 also our patient S83 was affected by adult visceral myopathy, a late onset condition characterized by fast progression and very poor outcome.
The further definition of the phenotypic spectrum associated with ACTG2 causing variants in larger CIPO series will improve diagnosis and our understanding of disease prognosis.
Finally, our results allow to retrospectively speculate about the nature of the primary defect in patients carrying ACTG2 variants. Indeed, most of our patients initially received an INDB diagnosis and consequently were regarded as affected by intestinal neuropathy. Similar to our patients, one of whom with CIPO+megacystis (S9) has specifically been reassessed showing a histological picture of visceral myopathy rather than visceral neuropathy, some histopathological studies of the myenteric and submucosal plexuses reported abnormalities of ganglion cells in ACTG2 variant carrying CIPO and MMIHS patients.8, 20 These results rule out the histopathological INDB picture, confirmed in the above S9 patient, as a hallmark for enteric neuropathy and/or a feature peculiar of intestinal innervations defects, suggesting enteric nervous system abnormalities may be secondary consequences of an impaired gut motility caused by ACTG2 variants and can occur either alongside, in parallel or subsequently to a primary non-enteric nervous system defect. This is of utmost importance for CIPO classification and confirms the need for extensive correlations between histopathological/clinical phenotypes and genetic defects in gastrointestinal motility disorders.
References
Knowles CH, De Giorgio R, Kapur RP et al: The London classification of gastrointestinal neuromuscular pathology: report on behalf of the Gastro 2009 International Working Group. Gut 2010; 59: 882–887.
De Giorgio R, Cogliandro RF, Barbara G, Corinaldesi R, Stanghellini V : Chronic intestinal pseudo-obstruction: clinical features, diagnosis, and therapy. Gastroenterol Clin North Am 2011; 40: 787–807.
Costa M, Fava M, Seri M et al: Evaluation of the HOX11L1 gene as a candidate for congenital disorders of intestinal innervation. J Med Genet 2000; 37: E9.
Gath R, Goessling A, Keller KM et al: Analysis of the RET, GDNF, EDN3, and EDNRB genes in patients with intestinal neuronal dysplasia and Hirschsprung disease. Gut 2001; 48: 671–675.
Pingault V, Girard M, Bondurand N et al: SOX10 variants in chronic intestinal pseudo-obstruction suggest a complex physiopathological mechanism. Hum Genet 2002; 111: 198–206.
Gargiulo A, Auricchio R, Barone MV et al: Filamin A is mutated in X-linked chronic idiopathic intestinal pseudo-obstruction with central nervous system involvement. Am J Hum Genet 2007; 80: 751–758.
Lehtonen HJ, Sipponen T, Tojkander S et al: Segregation of a missense variant in enteric smooth muscle actin g-2 with autosomal dominant familial visceral myopathy. Gastroenterology 2012; 143: 1482–1491.
Holla OL, Bock G, Busk OL, Isfoss BL : Familial visceral myopathy diagnosed by exome sequencing of a patient with chronic intestinal pseudo-obstruction. Endoscopy 2014; 46: 533–537.
Thorson W, Diaz-Horta O, Foster J et al: De novo ACTG2 variants cause congenital distended bladder, microcolon, and intestinal hypoperistalsis. Hum Genet 2014; 133: 737–742.
Wangler MF, Gonzaga-Jauregui C, Gambin T et al: Heterozygous de novo and inherited variants in the smooth muscle actin (ACTG2) gene underlie megacystis-microcolonintestinal hypoperistalsis syndrome. PLoS Genet 2014; 10: e1004258.
Klar J, Raykova D, Gustafson E et al: Phenotypic expansion of visceral myopathy asso'ciated with ACTG2 tandem base substitution. Eur J Hum Genet 2015; 23: 1679–1683.
Tuzovic L, Tang S, Miller RS et al: New Insights into the genetics of fetal megacystis: ACTG2 variants, encoding γ-2 smooth muscle actin in megacystis microcolon intestinal hypoperistalsis syndrome (Berdon syndrome). Fetal Diagn Ther 2015; 38: 296–306.
Fava M, Borghini S, Cinti R et al: HOX11L1: a promoter study to evaluate possible expression defects in intestinal motility disorders. Int J Mol Med 2002; 10: 101–106.
Martucciello G, Torre M, Pini Prato A et al: Associated anomalies in intestinal neuronal dysplasia. J Pediatr Surg 2002; 37: 219–223.
Gauthier J, Ouled Amar Bencheikh B, Hamdan FF et al: A homozygous loss-of-function variant in MYH11 in a case with megacystis-microcolon-intestinal hypoperistalsis syndrome. Eur J Hum Genet 2015; 23: 1266–1268.
Mc Laughlin D, Puri P : Familial megacystis microcolon intestinal hypoperistalsis syndrome: a systematic review. Pediatr Surg Int 2013; 29: 947–951.
Guo DC, Pannu H, Tran-Fadulu V et al: Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet 2007; 39: 1488–1493.
Milewicz DM, Østergaard JR, Ala-Kokko LM et al: De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am J Med Genet 2010; 152: 2437–2443.
Richer J, Milewicz DM, Gow R et al: R179H mutation in ACTA2 expanding the phenotype to include prunebelly sequence and skin manifestations. Am J Med Genet 2012; 158A: 664–668.
Puri P, Shinkai M : Megacystis microcolon intestinal hypoperistalsis syndrome. Semin Pediatr Surg 2005; 4: 58–63.
Acknowledgements
We thank Giuseppe Santamaria for his technical support, and all the patients and families for participating in this study. We would also like to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. This work was supported by the Italian Ministry of Health through, both grant RF-2010–2314356, ‘Cinque per mille’ and Ricerca Corrente to the Gaslini Institute; a research grant from the Shenzhen Municipal Government of China (NO.CXZZ20130517144604091) and the Institutional Development Funds to Dr Hakon Hakonarson (Center for Applied Genomics at the Children’s Hospital of Philadelphia).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Matera, I., Rusmini, M., Guo, Y. et al. Variants of the ACTG2 gene correlate with degree of severity and presence of megacystis in chronic intestinal pseudo-obstruction. Eur J Hum Genet 24, 1211–1215 (2016). https://doi.org/10.1038/ejhg.2015.275
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.275
This article is cited by
-
Exploring the complexities of megacystis-microcolon-intestinal hypoperistalsis syndrome: insights from genetic studies
Clinical Journal of Gastroenterology (2024)
-
Use of whole genome sequencing to determine the genetic basis of visceral myopathies including Prune Belly syndrome
Journal of Rare Diseases (2023)
-
Multi-disciplinary Insights from the First European Forum on Visceral Myopathy 2022 Meeting
Digestive Diseases and Sciences (2023)
-
Recurrent ACTG2 gene variation in African degenerative visceral leiomyopathy
Pediatric Surgery International (2019)
-
Consanguinity and its relevance for the incidence of megacystis microcolon intestinal hypoperistalsis syndrome (MMIHS): systematic review
Pediatric Surgery International (2019)
