
Abstract
Noonan syndrome is a heterogeneous autosomal dominant disorder caused by mutations in at least eight genes involved in the RAS/MAPK signaling pathway. Recently, RIT1 (Ras-like without CAAX 1) has been shown to be involved in the pathogenesis of some patients. We report a series of 44 patients from 30 pedigrees (including nine multiplex families) with mutations in RIT1. These patients display a typical Noonan gestalt and facial phenotype. Among the probands, 8.7% showed postnatal growth retardation, 90% had congenital heart defects, 36% had hypertrophic cardiomyopathy (a lower incidence compared with previous report), 50% displayed speech delay and 52% had learning difficulties, but only 22% required special education. None had major skin anomalies. One child died perinatally of juvenile myelomonocytic leukemia. Compared with the canonical Noonan phenotype linked to PTPN11 mutations, patients with RIT1 mutations appear to be less severely growth retarded and more frequently affected by cardiomyopathy. Based on our experience, we estimate that RIT1 could be the cause of 5% of Noonan syndrome patients. Because mutations found constitutionally in Noonan syndrome are also found in several tumors in adulthood, we evaluated the potential contribution of RIT1 to leukemogenesis in Noonan syndrome. We screened 192 pediatric cases of acute lymphoblastic leukemias (96 B-ALL and 96 T-ALL) and 110 cases of juvenile myelomonocytic leukemias (JMML), but detected no variation in these tumoral samples, suggesting that Noonan patients with germline RIT1 mutations are not at high risk to developing JMML or ALL, and that RIT1 has at most a marginal role in these sporadic malignancies.
Similar content being viewed by others

Introduction
Noonan syndrome (NS) is a common autosomal dominant disorder clinically defined by a constellation of anomalies, of which facial dysmorphism is the most consistent. NS is associated with several partially penetrant developmental anomalies, such as postnatal growth retardation and failure to thrive, congenital heart defects, hypertrophic cardiomyopathy (HCM), hyperkeratosis, and hypotrichosis. NS patients show an increased risk of learning disabilities and intellectual deficiency. They also have an increased risk of developing several types of childhood malignancies,1, 2, 3 including acute leukemia, myeloproliferative disorders (MPDs) and juvenile myelomonocytic leukemia (JMML).4, 5
NS is caused by a dysregulation of the RAS/MAPK signaling pathway. The first clue to this pathophysiology was the identification of gain-of-function mutations in the PTPN11 gene (OMIM 176876) in 40% of NS patients.6 Several other genes involved in the RAS/MAPK cascade were later found to explain smaller subgroups of NS, with loose genotype–phenotype correlations: KRAS (OMIM 190070), SOS1 (OMIM 182530), RAF1 (OMIM 164760), NRAS (OMIM 164790), SHOC2 (OMIM 602775), and CBL (OMIM 165360).7 RIT1 (Ras-like without CAAX 1; OMIM 609591) was identified in 2013,8 and more recently, mutations in RRAS9 (OMIM 165090), RASA210 (OMIM 601589), SOS211, 12 (OMIM 601247), and LZTR111 (OMIM 600774) have added further heterogeneity to the NS landscape. Rarely, NS has also been reported in patients with mutations in the CFC (cardiofaciocutaneous syndrome) genes BRAF,11 MAP2K1, and MAP2K2.12 Taken together, mutations in all these genes explain probably roughly 70% of clinically convincing NS cases.6
The identification of RIT1 as a causative gene for NS was carried out using exome sequencing by Aoki et al.8 in 2013. They found mutations in this gene in 4 out of 14 patients screened by exome sequencing, and confirmed this finding by identifying by Sanger sequencing 13 further cases in a cohort of 166 individuals who were negative for previously known NS genes. The involvement of RIT1 mutations in some cases of NS was subsequently confirmed in 14 other patients by others.12, 15, 16
RIT1 is a widely expressed small GTPase belonging to a subfamily of the RAS family. It has 50% sequence homology with classic RAS GTPases (KRAS, HRAS, NRAS), and shows canonical GTPase activity but lacks the prenylation motif (CAAX, XXCC or CXC) required for its association with the plasma membrane.17 Similar to all RAS family GTPases, it contains five conserved amino-acid motifs (G1–G5) involved in phosphate binding (G1 and G3), GTP binding and hydrolysis (G4 and G5), and effector protein binding (G2). Alternate splicing results in multiple transcript variants. Variant 1 (RefSeq accession number NM_001256821.1) encodes the longest isoform counting 236 amino acids. Variant 2 encodes the reference isoform of 219 amino acids (NM_006912.5), in which translation is initiated at a downstream start codon located in exon 2. Functional differences between the two isoforms have not been investigated. The activity of RIT1 is suspected of being partially redundant with that of other RAS genes, a hypothesis that is now supported by its implication in NS. RIT1 is involved in the stress-mediated activation of the scaffolded prosurvival signaling complex p38-MK2-HSP27-AKT, a critical component of the cellular survival mechanism in response to stress.18 RIT1 is also involved in the activation of the EPHB2 and MAPK14 pathways upon nerve growth factor signaling, and promotes neuronal development and regeneration.17 Although RIT1 may not have a major role in normal RAS-ERK pathway activation, the expression of mutant RIT1 alleles in heterologous cells increases MAPK-ERK pathway activation10 or ELK transactivation,8 demonstrating a gain-of-function effect and further supporting a causative role for RIT1 in NS pathogenesis.
Interestingly, somatic mutations in RIT1 have also been reported recently in several malignancies including lung adenocarcinoma,19, 20, 21 hepatoblastoma,22 urinary tract carcinoma,23 and adult myeloid malignancies.24 Among the latter, somatic RIT1 mutations have most often been found in chronic myelomonocytic leukemia, a myelodysplastic/myeloproliferative neoplasm of adulthood sharing numerous features with JMML. Taken together with the report of an NS patient carrying a RIT1 mutation who developed ALL in childhood,8 this raised the possibility that germline RIT1 mutations could confer susceptibility to leukemia.
Here we report clinical data for 44 patients from 30 families carrying a germline RIT1 mutation. In parallel, we screened tumoral DNA from 302 cases of pediatric acute lymphoblastic leukemia (ALL) and JMML to evaluate the role of RIT1 mutation in childhood leukemogenesis.
Materials and methods
Patient selection
Our institution is the only one in France to screen for all known NS genes. Since 2002, we have collected a large series of samples sent to us for the diagnosis of RASopathies. Most samples (>90%) were collected through a network of clinical geneticists throughout France. Pediatric endocrinologists or cardiologists selected the remaining cases. Over the years, we have built up a series of patients with typical NS, who are systematically investigated for each new gene to be identified.
In the present study, we screened 500 patients referred to our lab for RASopathies. These patients were analyzed by two modalities. The first group consisted of 117 strictly selected patients, who screened negative for mutations in the whole coding regions of NS, CFC, and Costello genes: PTPN11, SOS1, RAF1, MAP2K1, MAP2K2, BRAF, CBL, SHOC2, NRAS, KRAS, and HRAS. Irrespective of the referring clinician’s opinion, expert clinicians from our Department re-evaluated all patient records and clinical photographs. All had a typical NS and fulfilled the Van der Burgt criteria.25 The second set consisted of 383 consecutive samples from patients suspected of having NS by our referees, analyzed prospectively without re-evaluation by our team. The set of tested genes was variable (data not shown), but all had PTPN11 and RIT1 sequenced. Written informed consent for genetic investigation was obtained from all patients or their parents. Clinical data were compiled by analyzing photographs of the patients together with a questionnaire, filled out by the referring clinician, containing 72 clinical items regarding neonatal data, characteristic facial features, heart defects, skin abnormalities, growth retardation, developmental delays or mental retardation, and the occurrence of a solid tumor or leukemia.
Screening for RIT1 was also performed in 302 samples of childhood hematopoietic malignancies, including the French cohort of JMML (n=110), B-cell lineage ALL (n=96) and T-ALL (n=96). ALL samples were collected through the EORTC-CLG 58 951 study.
Gene screening
Genotyping was performed on genomic DNA by bidirectional Sanger sequencing of exons and their flanking intron–exon boundaries. The complete RIT1 sequence, consisting of six exons, was obtained for 212 patients suspected of NS (117 patients of group 1 and the first 95 patients of group 2) and all leukemia samples. The remaining 288 patients of the second NS group were only screened for exons 2–5, in a routine setting, as no mutation was ever evidenced in other exons. Direct sequencing of PCR products was performed using the Big Dye Terminator Cycle Sequencing Ready Reaction Kit (ABI, Foster City, CA, USA). Reaction products were run on an automated capillary sequencer (ABI 3130 Genetic Analyzer; ABI). Sequences were aligned using Seqscape analysis software (ABI) and compared with reference sequences for genomic DNA and mRNA. RIT1 mutations were named according to the NCBI reference transcript sequence NM_006912.5 coding isoform 2. Previous implication of the mutations in cancer was checked by consulting the COSMIC Catalog for Somatic Mutations in Cancer (http://www.sanger.ac.uk/genetics/CGP/cosmic). Previous report of single-nucleotide variants was verified by consulting the Ensembl genome browser (http://www.ensembl.genome.org). The prediction of the effects of amino-acid substitutions on the function or structure of proteins was carried out by interspecies alignment and by using dedicated prediction software: SIFT (http://sift.jcvi.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), and MutationTaster (http://www.mutationtaster.org/). Paternity was confirmed by simple tandem repeat genotyping, using the PowerPlex 16 System (Promega, Fitchburg, WI, USA). GTPase protein sequences were aligned using ClustalW (http://www.ebi.ac.uk/Tools/msa/clustalw2/) (Figure 1). All variants were declared in the ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar/) with patient accession numbers SCV000211874–SCV000211887.

Protein sequence alignment of human RIT1, RIT2, HRAS, NRAS, KRAS and RRAS small GTPases. The G1–G5 domains and switch I and II regions are indicated by blue and green bars, respectively. Gray arrows indicate the oncogenic Gly12, Gly13, and Gln61 mutation hotspots. Red arrows indicate amino acids where novel RIT1 germline mutations were identified in this study. Amino acids where RIT1 germline8 or somatic mutations in hematopoietic malignancies (from (Gomez-Segui et al.24 and Cosmic database) were identified in previous studies are indicated by orange arrows and orange asterisk, respectively. In contrast with what is found in canonical RAS proteins (H/N/KRAS), (i) RIT1 mutations cluster in the switch regions and (ii) germline and oncogenic somatic are overlapping. A full color version of this figure is available at the European Journal of Human Genetics journal online.
Results
We found 14 distinct RIT1 mutations in 30/500 probands (first group: 27/117; second group: 3/383) suspected of having NS (Table 2). Among them, three were novel amino-acid substitutions. The p.(Ala77Thr), found twice, occurred de novo in one patient and was inherited from an affected parent in the second. The p.(Ala84Val) variant cosegregated with the phenotype in a mother and her two affected children. Paternal sample was not obtained for the patient carrying the p.(Asp51Tyr). The three variants were located into regions that were evolutionary conserved, highly homologous with other RAS family members (Figure 1),8 and predicted to be damaging by three bioinformatic algorithms (Table 1). The other 11 variants had already been identified as pathogenic in previous reports (Table 2).
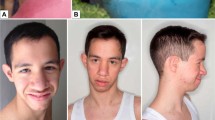
In eight sporadic probands, mutations occurred de novo. For the 13 remaining sporadic probands, at least one parent sample was not available for genotyping. For nine probands, a mutation was also identified in at least one relative: in six cases, the mutation was found in one parent or one child, in three families, there were three affected subjects in two generations. All relatives considered as being clearly or mildly affected on clinical grounds before genotyping were found to carry a mutation. The mutation was inherited from the mother in 9 out of 14 genetically confirmed transmission. Clinical appearance of patients with RIT1 mutations is shown in Figure 2. Table 3 summarizes the clinical data of our 44 patients, compared with data available from 4 recent publications, and data from patients with NS and PTPN11 mutations.26 Clinical information was limited for some affected parents, as they had a mild phenotype for which further clinical investigation on a routine basis was thought to be unjustified (most of them were undiagnosed until the identification of NS in the proband).

Clinical appearance of patients with RIT1 mutations. (a) Female patient in infancy and at 7 years of age with c.244T>C (p.Phe82Leu), (b) daughter (at birth and at 9 years of age) and mother with c.104G>C (p.Ser35Thr), (c) adult patient with c.242A>G (p.Glu81Gly), and (d) daughter (aged 4 years) and mother with c.284G>C (p.Gly95Ala).
About one-third of patients displayed prenatal symptoms. Neonatal growth parameters were normal. Poor feeding was noted in roughly half of the cases in infancy, but no patients required tube feeding. Only 4/33 patients (12%) had a stature of <−2 SD, and their median height was only −0.45 SD One of the probands had a height of +2 SD at the age of 8 years. Speech delay was observed in 13/30 (43%) of patients over 18 months, learning difficulties in 9/23 (39%) over age 5 years, but among those, only 4/23 (17%) required special education. One of the affected adults completed law school. Contrary to the observation of Aoki et al.,8 HCM was present in only 13/38 (34%) of our probands (including two patients having only septal hypertrophy), whereas pulmonary stenosis or pulmonary valve dysplasia was present in 29/38 probands (76%). Skin anomalies were uncommon and usually mild, but some patients exhibited sparse eyebrows (10%), hyperkeratosis (11%), and deep palmar creases (38%). Café-au-lait spots were unexpectly found in four patients. None of our patients would have been diagnosed with CFC syndrome. At least three patients presented with severe prenatal lymphatic involvement. Four patients had hepato- and/or splenomegaly, one of whom had thrombocytopenia; another had thrombocytemia, while a third developed JMML. This latter patient was a premature newborn (34 WG) who had macrosomia, HCM, pleural effusion, and normal blood count at birth. At day 17, while in neonatal intensive care unit, he developed hyperleucocytosis (WBC: 76.4 × 109/l; monocytes: 24.4 × 109/l) with morphological evidence of bone marrow progenitors and undifferentiated myeloid blasts in peripheral blood (Figure 3). Diagnosis of JMML was confirmed by in vitro endogenous growth of myeloid progenitors. He died at 1 month of age of multivisceral failure. He carried the recurrent p.Gly95Ala change. No other patient developed a hematological malignancy or any other tumor over a median follow-up period of 10 years and 4 months.

May–Grünwald–Giemsa-stained blood smear at D17 of life demonstrating JMML in a patient with RIT1 p.Gly95Ala. Morphological evidence of bone marrow progenitors in peripheral blood together with excess of undifferentiated myeloid blasts and monocytosis (x100 magnification). Red arrowhead shows an undifferentiated myeloid blast, whereas black arrowhead and black asterisk indicate a myelocyte and an acidophilic erythroblast respectively. Green arrowhead indicates a monocyte. A full color version of this figure is available at the European Journal of Human Genetics journal online.
No other pathogenic RIT1 mutation could be detected among the 110 sporadic JMML or 192 childhood ALL samples (96 T-cell lineage ALL, 96 B-cell precursor ALL) that were tested. The upper limit of the confidence interval of the observed proportion in ALL (0/192 samples) is 0.019 (Clopper–Pearson’s exact method and Wilson score). Hence, RIT1 mutations should be present at most in 2% of ALL.
Discussion
RIT1 has recently emerged as a new player in NS. However, few patients with pathogenic RIT1 mutations have been described so far. In their cohort of 180 NS patients previously screened for known NS genes, Aoki et al.8 uncovered 17 unrelated patients with 8 different RIT1 mutations Among eight patients for whom the parents were genotyped, only one case was inherited, from a mother whose clinical phenotype was not described. Fifteen patients fulfilled the Van der Burgt criteria and two had a clinical diagnosis of CFC syndrome. Three additional series with a further 15 patients have recently been added. Bertola et al.15 have described six Brazilian patients picked up by exome sequencing from a cohort of 70 NS patients who screened negative for PTPN11, SOS1, KRAS, RAF1, SHOC2, and CBL. Gos et al.16 have identified four RIT1-NS cases out of 106 patients without mutations in PTPTN11, SOS1, or RAF1, whereas Chen et al.10 have identified five patients with RIT1 mutations after exome sequencing of 25 patients.10 In the present study, we identified 30 additional probands and 14 relatives with NS carrying a RIT1 mutation, raising the total number of reported cases to 76. Three variants predicted to be pathogenic in silico were not reported before. All were located in the functional regions were all pathogenic variants cluster and targeted amino acids remarkably conserved among GTPAses (Table 1 and Figure 1). p.(Ala77Thr) was found in two unrelated probands, once transmitted by an affected parent, and once de novo, and the p.(Ala84Val) variant cosegregated with the phenotype in three patients and two generations, making them convincing pathogenic variants. Pathogenicity remains putative for p.(Asp51Tyr). However, transforming capacities have been demonstrated for a closely related variant (p.Asp51Val) substitution previously found in a patient with chronic myelomonocytic leukemia.19
Our findings confirm the tight clustering of RIT1 mutations (Figure 1). Indeed, in contrast with RAS genes, all RIT1 mutations described so far in either NS or malignancies24 are highly clustered in the G1–G3 and switch II regions, that is, the most highly conserved part of RIT1 when aligned with RAS proteins,8, 27 and also among species. The C-terminal end of human RIT1 is highly heterologous as compared with other RAS family members. Strikingly, no proven deleterious mutation has been reported so far in regions that are not both evolutionarily conserved and homologous to other RAS family proteins.8, 19
Altogether, RIT1 appears to be a new minor NS gene. In our study, the percentage of mutants was strikingly different between the two sets of patients. A pathogenic RIT1 mutation was found in 27 (23%) of the strictly selected first set of 117 patients with very convincing NS features, but in only 3 of the 383 (0.8%) loosely selected NS cases. In the latter cohort, the frequency of PTPN11 mutations is about 20%, which is half that of the 41% frequency expected in more carefully selected patients.6 Consequently, the frequency of RIT1 mutations can be roughly estimated to be around 2–3%, which ranks RIT1 at the same level as KRAS in the spectrum of NS genes. The high frequency of RIT1 mutations in strictly selected NS patients and the low frequency in more loosely selected patients is consistent with the fact that these patients usually display a typical NS phenotype, that is, a phenotype similar to that of patients with PTPN11 mutations. In Aoki’s report, two patients were initially diagnosed as CFC. One of these patients has no ID at the age of 4 years (making retrospectively a clinical diagnosis of CFC challenging). The second one has a more severe delay (DQ of 44 at 23 months) but a mild craniofacial phenotype (see Aoki’s Figures 1a and d). Interestingly, the two causative mutations (p.Met90Ile and p.Phe82Leu) were subsequently found in patients clinically diagnosed as NS (by Gos et al.16 and by ourselves), illustrating the well-known challenge of intrinsic variability of most RASopathy mutations, and the limits of clinically based differential diagnosis of NS and CFC.
Surprisingly, the comparison of the phenotypic profiles of RIT1 and PTPN11 was difficult, as few series with detailed clinical data are available. Using the literature data compiled on PTPN11-NS by Sarkozy et al.,26 we conclude that RIT1-NS patients have a typical NS face, but are significantly taller. The incidence of pulmonary stenosis and septal defects is similar between the two genotypes, but HCM is roughly five times more frequent with RIT1 (45% vs 9%). The incidence of developmental delay (in a broad sense) in the reported RIT1-NS cases is ~40%. Only 17% have intellectual deficiency requiring special education. Because of a common confusion among learning difficulties, motor delay, speech delay with normal IQ, and true cognitive deficiency in the literature, it is difficult to evaluate whether RIT1-NS cases are – or are not – less likely to display intellectual deficiency than PTPN11-NS cases. The impact of RIT1 mutations on growth is significantly milder than that of PTPN11 mutations (26% vs 76%). Similarly, cryptorchidism and pectus deformities appear significantly less common with RIT1 mutations. Whereas males are slightly more common in patients with PTPN11 mutations,5 the combined sex ratio of the 70 RIT1 patients is 0.75, reflecting an unexpected tilt towards females (proportion of males: 0.43; confidence interval at P=0.05: 0.32–0.56). To summarize, although RIT1 mutations are not associated with a specific recognizable phenotype among the RASopathies, their presentation differs somewhat from the more common PTPN11-related cases.
Our patients differ in some aspects from previous series. In particular, they appear to display a milder phenotype. The different modes of screening/selection could explain the fact that, in previous publications, the patients shared a more severe and internally consistent phenotype: in previous series, RIT1 was identified by exome screening in many patients. Selection criteria may have been more rigorous for exome cases, biasing the panel toward severe or ‘very typical cases’ that exceed the Van der Burgt criteria. In our series, group 1 was similarly selected towards ‘typical cases’, whereas group 2 represented a routine diagnostic setting. We also identified a much higher incidence of familial cases than other groups. This is consistent with the fact that patients with RIT1 mutations have few general or cognitive problems and are thus likely to show greater reproductive fitness. Interestingly, RIT1 mutations share some rare complications with other RASopathy genes: JMML, aggressive giant cell tumor of the jaw,28 and autoimmune disorders (Graves disease and lupus erythematosus).29
RIT1 and cancer
NS patients in general have an increased risk of developing several types of childhood malignancies,1, 2, 3 including acute leukemia or MPDs such as JMML.5 We report here the first patient with JMML and a constitutional RIT1 mutation, reinforcing the pathophysiological link between the two diseases. This observation provides a new example of the leukemogenic role of small GTPases that do not belong to the core RAS-MAPK signaling cassette. Indeed, we have recently shown that RRAS, another RAS-like GTPase, is mutated in rare cases of NS and in some cases of JMML.9 Similar to classic Ras and R-Ras, Rit proteins have been shown to display oncogenic properties, and GTPase-deficient variants cause growth transformation in NIH 3T3 mouse fibroblasts.30 The expression of wild-type or oncogenic RIT1 in PC6 cells induces in most cases both the phosphorylation of ERK via MEK activation and a robust activation of PI3K/AKT signaling.19 In line with these in vitro findings, somatic missense variants and small in-frame insertions/deletions targeting the switch II domain of RIT1 have recently be found by exome sequencing in 10/413 (2.4%) lung adenocarcinomas.19, 20, 21 RIT1 mutations were also identified in various adulthood malignancies including myeloproliferative and mixed myelodysplastic/myeloproliferative neoplasms such as chronic myelomonocytic leukemia, an adult neoplasm resembling JMML.23, 24 Although RIT1 mutations were only reported in adult malignancies so far, one of Aoki's cases survived an ALL diagnosed at age 5. No other patient described so far including ours developed cancer but the total number of patients identified with a RIT1 mutation and the median age of follow-up remains low. We thus used a ‘second entrance’ by screening unselected tumor cohorts for RIT1 mutations. We focused on 2 specific leukemia: ALL because the first and only case of tumor reported so far in RIT1 associated NS was an ALL, and JMML because of our patient described above, and more generally because this rare leukemia is specifically associated with RASopathies. The statistical power of these analyses remains limited. However both (1) the lack of childhood ALL in our series of NS patients with RIT1 mutation and (2) the lack of RIT1 mutations in a series of about 200 childhood ALL of B or T-lineage, reinforce each other, and do not support the possibility that RIT1 patients are more prone than other NS patients to developing JMML or childhood ALL of the B- and T-cell lineages. Together with other reports, our data suggest that germline RIT1 mutations are probably not strongly oncogenic in childhood. We have suggested to follow WBC every 6 months in children with any form of NS up to the age of 2 years to detect early increased lymphomonocytosis.5 This would also apply to RIT1-NS patients, but no other specific clinical follow-up has to be proposed. In addition, our data suggest that somatically acquired RIT1 mutations have at most a marginal role in these sporadic malignancies of childhood.
Oncogenic RIT1 mutations apparently show no tissue specificity as they can be shared by different tumor types. Moreover, unlike what has previously been reported for KRAS, BRAF, and PTPN11, the spectrum of pathogenic somatic RIT1 mutations evidenced in tumors overlaps with that of germline mutations found in patients with NS (Table 1). This raises the concern that patients harboring RIT1 mutations may be prone to some forms of cancer, although our finding that RIT1 mutations are not commonly observed in JMML and childhood ALL suggests that its tumor spectrum could be different from that of other RASopathy genes. This issue remains open since very few RIT1-NS patients have been identified so far and most of them are still children, but it certainly deserves close attention.
In conclusion, we confirm here the phenotype of RIT1-related NS in 30 new index patients and 14 relatives. RIT1 represents a minor etiology for NS, explaining about 3% of cases. Although these patients are not individually recognizable as RIT1-NS, their phenotype appears to be characteristic in terms of facial features but relatively mild in other respects, with few cognitive or growth-related deficits, but a higher incidence of HCM, and a risk of JMML. No other specific cancer risk could be associated with this form of NS so far.
References
Kratz CP, Rapisuwon S, Reed H, Hasle H, Rosenberg PS : Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am J Med Genet C 2011; 157: 83–89.
Hasle H : Malignant diseases in Noonan syndrome and related disorders. Horm Res 2009; 72 (Suppl 2): 8–14.
Kratz CP, Franke L, Peters H et al: Cancer spectrum and frequency among children with Noonan, Costello, and cardio-facio-cutaneous syndromes. Br J Cancer 2015; 112: 1392–1397.
Bader-Meunier B, Tchernia G, Mielot F et al: Occurrence of myeloproliferative disorder in patients with Noonan syndrome. J Pediatr 1997; 130: 885–889.
Strullu M, Caye A, Lachenaud J et al: Juvenile myelomonocytic leukaemia and Noonan syndrome. J Med Genet 2014; 51: 689–697.
Romano AA, Allanson JE, Dahlgren J et al: Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics 2010; 126: 746–759.
Rauen KA : The RASopathies. Annu Rev Genomics Hum Genet 2013; 14: 355–369.
Aoki Y, Niihori T, Banjo T et al: Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am J Hum Genet 2013; 93: 173–180.
Flex E, Jaiswal M, Pantaleoni F et al: Activating mutations in RRAS underlie a phenotype within the RASopathy spectrum and contribute to leukaemogenesis. Hum Mol Genet 2014; 23: 4315–4327.
Chen PC, Yin J, Yu HW et al: Next-generation sequencing identifies rare variants associated with Noonan syndrome. Proc Natl Acad Sci USA 2014; 111: 11473–11478.
Yamamoto GL, Aguena M, Gos M et al: Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J Med Genet 2015; 52: 413–421.
Cordeddu V, Yin JC, Gunnarsson C et al: Activating Mutations Affecting the Dbl Homology Domain of SOS2 Cause Noonan Syndrome. Hum Mutat 2015; 36: 1080–1087.
Lee BH, Kim JM, Jin HY, Kim GH, Choi JH, Yoo HW : Spectrum of mutations in Noonan syndrome and their correlation with phenotypes. J Pediatr 2011; 159: 1029–1035.
Nava C, Hanna N, Michot C et al: Cardio-facio-cutaneous and Noonan syndromes due to mutations in the RAS/MAPK signalling pathway: genotype-phenotype relationships and overlap with Costello syndrome. J Med Genet 2007; 44: 763–771.
Bertola DR, Yamamoto GL, Almeida TF et al: Further evidence of the importance of RIT1 in Noonan syndrome. Am J Med Genet A 2014; 164A: 2952–2957.
Gos M, Fahiminiya S, Poznanski J et al: Contribution of RIT1 mutations to the pathogenesis of Noonan syndrome: four new cases and further evidence of heterogeneity. Am J Med Genet A 2014; 164: 2310–2316.
Shi GX, Cai W, Andres DA : Rit subfamily small GTPases: regulators in neuronal differentiation and survival. Cell Signal 2013; 25: 2060–2068.
Cai W, Carlson SW, Brelsfoard JM et al: Rit GTPase signaling promotes immature hippocampal neuronal survival. J Neurosci 2012; 32: 9887–9897.
Berger AH, Imielinski M, Duke F et al: Oncogenic RIT1 mutations in lung adenocarcinoma. Oncogene 2014; 33: 4418–4423.
Imielinski M, Berger AH, Hammerman PS et al: Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012; 150: 1107–1120.
Cancer Genome Atlas Research N: Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014; 511: 543–550.
Li JT, Liu W, Kuang ZH, Zhang RH, Chen HK, Feng QS : Mutation and amplification of RIT1 gene in hepatocellular carcinoma. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2004; 21: 43–46.
Guo G, Sun X, Chen C et al: Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat Genet 2013; 45: 1459–1463.
Gomez-Segui I, Makishima H, Jerez A et al: Novel recurrent mutations in the RAS-like GTP-binding gene RIT1 in myeloid malignancies. Leukemia 2013; 27: 1943–1946.
van der Burgt I, Berends E, Lommen E, van BS, Hamel B, Mariman E : Clinical and molecular studies in a large Dutch family with Noonan syndrome. Am J Med Genet 1994; 53: 187–191.
Sarkozy AD, Marino MC, dallapicola B Genotype–phenotype correlations in Noonan syndrome; in: Zenker M (ed): Noonan Syndrome and Related Disorders. Basel: Karger, 2009, Vol 17, pp 40–54.
Lee CH, Della NG, Chew CE, Zack DJ : Rin, a neuron-specific and calmodulin-binding small G-protein, and Rit define a novel subfamily of ras proteins. J Neurosci 1996; 16: 6784–6794.
Beneteau C, Cave H, Moncla A et al: SOS1 and PTPN11 mutations in five cases of Noonan syndrome with multiple giant cell lesions. Eur J Hum Genet 2009; 17: 1216–1221.
Bader-Meunier B, Cave H, Jeremiah N et al: Are RASopathies new monogenic predisposing conditions to the development of systemic lupus erythematosus? Case report and systematic review of the literature. Semin Arthritis Rheum 2013; 43: 217–219.
Rusyn EV, Reynolds ER, Shao H et al: Rit, a non-lipid-modified Ras-related protein, transforms NIH3T3 cells without activating the ERK, JNK, p38 MAPK or PI3K/Akt pathways. Oncogene 2000; 19: 4685–4694.
Acknowledgements
The authors thanks the families for their collaboration and trust, and S Rasika for her help in improving the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Cavé, H., Caye, A., Ghedira, N. et al. Mutations in RIT1 cause Noonan syndrome with possible juvenile myelomonocytic leukemia but are not involved in acute lymphoblastic leukemia. Eur J Hum Genet 24, 1124–1131 (2016). https://doi.org/10.1038/ejhg.2015.273
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.273
This article is cited by
-
RIT1 Promotes Glioma Proliferation and Invasion via the AKT/ERK/NF-ĸB Signaling Pathway
Journal of Molecular Neuroscience (2022)
-
Advances in germline predisposition to acute leukaemias and myeloid neoplasms
Nature Reviews Cancer (2021)
-
Identifying facial phenotypes of genetic disorders using deep learning
Nature Medicine (2019)
