
Abstract
PEHO syndrome (OMIM no. 260565) is characterized by myoclonic jerking and infantile spasms, profound psychomotor retardation with the absence of motor milestones and speech, absence or early loss of visual fixation with atrophy of optic discs by 2 years of age and progressive brain atrophy on neuroimaging. We describe the results of a genomic study of a girl with PEHO syndrome and review the literature on cases with a disease-causing variant in the same gene. Exome sequencing of the index and unaffected parents followed by Sanger confirmation identified nine candidate genes harboring nonsynonymous rare variants identified by trio whole-exome sequencing. The de novo variant, a missense variant (c.296C>T, p.(T99M)), affecting the motor domain of KIF1A was considered the pathogenic mutation. The literature review revealed 24 cases with disease-causing variants in the motor domain of KIF1A, of which three met all the criteria for PEHO syndrome and an additional patient with incomplete clinical data met four of the five criteria. If the criteria were modified to include cases with any convulsive disorder and less profound intellectual disability, a total of six patients met all five of the criteria, three patients met four of the criteria and six met three of the criteria. Our results indicate that the molecular basis for PEHO syndrome, in at least a subset of patients, is a dominant KIF1A variant affecting the motor domain of the protein. Variable expressivity is seen with recurrent variants causing the full phenotype of PEHO syndrome in some patients and in other patients, a partial or milder PEHO phenotype.
Similar content being viewed by others


Introduction
PEHO syndrome (OMIM no. 260565), a condition first described in 1991 in 14 patients from 11 Finnish families,1 is diagnosed clinically based on the criteria published by Somer et al2 who performed an extensive review of the condition and established the necessary features of infantile hypotonia, convulsive disorder manifesting with myoclonic jerking and infantile spasms, profound psychomotor retardation with the absence of motor milestones and speech, absence or early loss of visual fixation with atrophy of optic discs by 2 years of age and progressive brain atrophy on neuroimaging studies, particularly in the cerebellum and brain stem with milder supratentorial atrophy.2 Supportive criteria for this diagnosis are subtle dysmorphic features (narrow forehead, epicanthic folds, short nose, open mouth, receding chin and tapered fingers), edema of the face and limbs, brisk tendon reflexes in early childhood, abnormal brain stem auditory-evoked potentials, absent cortical responses of somatosensory-evoked potentials, slow nerve conduction velocities in late childhood and dysmyelination on magnetic resonance imaging. Microcephaly is progressive and not usually present at birth.
Although originally described in Finnish families, PEHO syndrome has since been reported in patients of other ethnicities, both as sporadic and familial cases.3, 4 In 2011, we published findings pertaining to two unrelated girls with PEHO syndrome.3 Patient 1 met all the necessary criteria for this diagnosis and continues to be followed in our center. Patient 2 died at the age of 23 months and could not be included in this study owing to the lack of available DNA. We report the results of whole-exome sequencing (WES) of patient 1 and her unaffected non-consanguineous parents, which identified a de novo dominant variant in the motor domain of KIF1A. Based on a review of the clinical phenotype of previously published patients with the same as well as other de novo KIF1A disease-causing variants, we propose that in a subset of patients, the molecular basis for PEHO syndrome is a dominant KIF1A variant affecting the motor domain of the protein.
Materials and methods
As part of the TIDEX gene discovery project (UBC IRB approval H12-00067), WES was performed for the index (patient 1 in 2011 case report) and her unaffected parents using the Agilent SureSelect Kit and Illumina HiSeq 2000 (Perkin-Elmer, Santa Clara, CA, USA). The sequencing reads (~30 × coverage) were aligned to the human reference genome version hg19 and rare variants (MAF <0.01) were identified and assessed for their potential to disrupt protein function, and subsequently screened under a series of genetic models. The details of the filtering process can be found in the Supplementary Data (Supplementary Table S1 and S2). The genetic variant data of the disease-causing variant identified in this study was submitted to ClinVar (accession number SCV000243778; http://www.ncbi.nlm.nih.gov/clinvar/).
Previous publications on patients with variants affecting the motor domain of KIF1A were retrieved through searches of PubMed using KIF1A and Kinesin family member 1A as keywords (June 2015). Their clinical presentation was reviewed to determine if their features meet the diagnostic criteria of PEHO syndrome (Table 1), and analyzed based on available information to determine whether there might be a specific KIF1A genotype causing PEHO syndrome. All variants refer to positions according to NCBI RefSeq NM-001244008.1.
Results
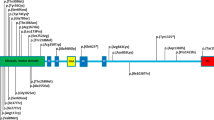
The clinical details of our patient were reported previously.3 She was diagnosed with PEHO syndrome based on infantile hypotonia; visual inattentiveness noted at an early age and bilateral optic atrophy diagnosed at 2 years of age; infantile spasms difficult to control with an EEG showing hypsarrhythmia from the age of 18 months; and profound developmental delay and an MRI of her brain at age 8 months showing a thin corpus callosum and small optic chiasm but repeat MRI at 2 years showing progressive severe loss of cerebellum, marked atrophy of the corpus callosum with dilatation of the lateral ventricle (Figure 1). She also had dysmorphic features that included bitemporal narrowing, down-slanted palpebral fissures, short nose, open mouth with curved upper lip and retrognathia (Figure 2). Of the nine candidate genes (eight with compound heterozygous variants and one with de novo heterozygous variant) identified by trio WES, the de novo variant, a missense variant c.296C>T (reference sequence NM_001244008.1), p.(T99M) in KIF1A located on chromosome 2, was classified as pathogenic according to recently published ACMG Standards and Guidelines.5 This variant has not been previously recorded in ExAC (Exome Aggregation Consortium), NHLBI ESP or our in-house genome database (comprising >350 WES and whole-genome sequencing (WGS) results) and has only been reported in dbSNP142 (rs387906799) in patients with cognitive impairment and evidence of progressive neurodegeneration.6, 7, 8, 9 In silico prediction tools, such as SIFT and Polyphen 2, predict this missense variant to be damaging, and the variant has a high (26.7) CADD (Combined Annotation Dependent Depletion) score.10 Sanger sequencing was performed in the affected individual and both unaffected parents and confirmed that the affected child was heterozygous for the variant, which was not present in either parent (Figure 3).

MRI of the brain showing prominent folia of the cerebellar hemispheres and vermis with increased surrounding subarachnoid cerebral spinal fluid indicative of severe symmetrical volume loss of both the cerebellar hemispheres and cerebellar vermis.

Patient at 3 years and 4 months of age. Photo previously published in Alfadhel et al.3

Pedigree and Sanger sequencing results.
Before this report, 24 patients in 22 families, including one set of monozygotic twins and one father and son pair, have been reported with de novo heterozygous variants affecting the motor domain of KIF1A protein. Lee et al8 reported on 14 of these patients and provided evidence of the functional impact of these variants. They concluded that the main clinical features seen in patients with these variants are: moderate to severe developmental delay, cerebellar atrophy on MRI with variable cerebral atrophy, optic nerve atrophy, progressive spasticity affecting mainly the lower limbs and peripheral neuropathy. The six patients reported by Nieh et al9 all presented with severe developmental delay, hypotonia and a degenerative neurologic course with progressive cerebral and cerebellar atrophy. Microcephaly, cortical visual impairment, optic neuropathy and epilepsy, all features of PEHO syndrome, were observed in some patients. Table 1 provides an overview of the data on all reported cases. Fourteen different de novo disease-causing variants were reported, and of the 25 patients (including our own), seven have the same variant observed in our index case. The c.757G>A (p.(E253K)) was observed in three patients, whereas the c.946C>T (p.(R316W)) variant in two. In addition, three patients were found to have a variant affecting the same residue: p.(R216P), p.(R216C) and p.(R216H). Based on the available clinical information in the published reports, there appears to be a variable expressivity of the recurrent variants. Of the seven patients with the c.296C>T (p.(T99M)) seen in our patient, only three patients meet the published clinical diagnostic criteria for PEHO syndrome (Table 1).
Although none of the previously reported carriers of KIF1A variants had been diagnosed with PEHO syndrome, a review of their clinical presentation indicates that three patients met the necessary criteria for this diagnosis. One patient met four of the criteria with no information available about visual acuity/optic atrophy. As most genetic disorders have a variable phenotype, if one was to expand the phenotype of PEHO syndrome to include patients with milder phenotypes and modify the criteria to include: any convulsive disorder as opposed to only myoclonic jerking and infantile spasms; any level of intellectual disability as opposed to only cases with profound intellectual disability; and any degree of optic atrophy; then two additional patients would meet all five of the criteria; two additional patients met four out of the five criteria; and a total of six patients, three out of five of the criteria. Only one variant affecting the motor domain of KIF1A, p.(S69L), seen in a father and son, resulted in pure spastic paraplegia. Progressive spasticity of the lower limbs with brisk reflexes was a feature seen in all but two patients with heterozygous variants affecting the motor domain of KIF1A.11 This feature, although not a diagnostic criteria of PEHO syndrome, was described in the cases reported by Salonen et al1 and Somer et al.2 As mentioned above, PEHO syndrome patients, including our patient, present with subtle dysmorphic features and edema of the face and limbs, especially in early childhood. Such findings were not mentioned in the patients previously reported with heterozygous variants in the KIF1A gene.
Discussion
PEHO syndrome has been assumed to be an autosomal recessive condition based on the report by Somer et al2 of 19 affected children in 14 Finnish families and one report of PEHO in two Japanese siblings.4 However, no known parental consanguinity was reported in any of the reported cases of PEHO syndrome. In the Finnish families, two mothers of affected children were fourth cousins but the authors point out that most grandparents’ birth places did not cluster into a few regions, which is the case in several other hereditary disorders typical of the Finns. This raises the possibility that some of the recurrences reported may be because of germline mosaicism or somatic mosaicism in one parent, and that PEHO syndrome is etiologically heterogeneous. The latter may explain why, even among Finnish cases, it has been difficult to find the gene responsible for this condition.
De novo disease-causing variants in KIF1A show phenotypic variability even in patients with the same variants (Table 1). The present study shows evidence that, in some patients, a variant affecting the motor domain of KIF1A causes the full phenotype of PEHO syndrome, and in other patients, a partial or milder PEHO phenotype. Seizures appear to be the least consistent finding, whereas the development of spasticity of the lower limbs is the most consistent finding as children age.
The clinical features seen in PEHO syndrome are found in several conditions characterized by early-onset global developmental delay, encephalopathy, optic atrophy, edema±cerebellar and cerebral atrophy. A literature search (OMIM and PubMed) yielded the following differential diagnosis: Aicardi syndrome, SCN2A encephalopathy, mevalonic aciduria, the carbohydrate-deficient glycoprotein syndromes, mitochondrial diseases due to nuclear as well as mtDNA variants, and – if especially brain atrophy is present – autosomal recessive cerebellar hypoplasia, Joubert syndrome, and olivo-pontine cerebellar atrophies (Orphanet 2836). The inheritance pattern of these conditions varies, making it extremely important to determine the underlying genetic disease-causing variant. Based on our findings, we propose molecular analysis of KIF1A in all patients who present with a combination of infantile hypotonia, developmental disability, visual impairment with optic atrophy and cerebellar atrophy. Confirmation of de novo KIF1A variants affecting the motor domain of the protein in patients clinically diagnosed with PEHO syndrome would allow for appropriate genetic counseling of the parents, and is a first step to unraveling the pathophysiology of this detrimental condition. Finally, the genetic heterogeneity of PEHO syndrome can be delineated by WES/WGS analysis of all KIF1A-negative patients meeting the criteria as defined more than 20 years ago.
References
Salonen R, Somer M, Haltia M et al: Progressive encephalopathy with edema, hysparrhythmia, and optic atrophy (PEHO syndrome). Clin Genet 1991; 39: 287–293.
Somer M : Diagnostic criteria and genetics of the PEHO syndrome. J Med Genet 1993; 30: 932–936.
Alfadhel M, Yong SL, Lillquist Y, Langlois S : Precocious puberty in two girls with PEHO syndrome: a clinical feature not previously described. J Child Neurol 2011; 26: 851–857.
Fujimoto S, Yokochi K, Nakano M, Wada Y : Progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy (PEHO syndrome) in two Japanese siblings. Neuropediatrics 1995; 26: 270–272.
Richards S, Aziz N, Bale S et alACMG Laboratory Quality Assurance Committee: Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–424.
Hamdan FF, Gauthier J, Araki Y et al: Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am J Hum Genet 2011; 88: 306–316.
Okamoto N, Miya F, Tsunoda T et al: KIF1A mutation in a patient with progressive neurodegeneration. J Hum Genet 2014; 59: 639–641.
Lee JR, Srour M, Kim D et al: De novo mutations in the motor domain of KIF1A cause cognitive impairment, spastic paraparesis, axonal neuropathy, and cerebellar atrophy. Hum Mutat 2015; 36: 69–78.
Nieh SE, Madou MRZ, Sirajuddin M et al: De novo mutations in KIF1A cause encephalopathy and brain atrophy. Ann Clin Transl Neurol 2015; 2: 623–635.
Kircher M, Witten DM, Jain P et al: A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014; 46: 310–315.
Ylikallio E, Kim D, Isohanni P et al: Dominant transmission of de novo KIF1A motor domain variant underlying pure spastic paraplegia. Eur J Hum Genet 2015; 23: 1427–1430.
Acknowledgements
We gratefully acknowledge the family for their participation in this study; Dr M Alfadhel for the detailed clinical description; Mrs X Han for Sanger sequencing; Mrs M Higginson for gDNA extraction, sample handling and technical data (University of British Columbia, Vancouver, BC, Canada). This work was supported by funding from Genome BC (SOF-195 grant) and the Canadian Institutes of Health Research (No. 301221 grant); informatics infrastructure was supported by Genome BC and Genome Canada (ABC4DE Project). CvK is a Michael Smith Foundation for Health Research Scholar. CJDR is supported by a CIHR New Investigator award.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Langlois, S., Tarailo-Graovac, M., Sayson, B. et al. De novo dominant variants affecting the motor domain of KIF1A are a cause of PEHO syndrome. Eur J Hum Genet 24, 949–953 (2016). https://doi.org/10.1038/ejhg.2015.217
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.217
This article is cited by
-
Neurogenetic disorders across the lifespan: from aberrant development to degeneration
Nature Reviews Neurology (2022)
-
Monoallelic KIF1A-related disorders: a multicenter cross sectional study and systematic literature review
Journal of Neurology (2022)
-
KIF1A-related autosomal dominant spastic paraplegias (SPG30) in Russian families
BMC Neurology (2020)
