
Abstract
AIMP1/p43 is a multifunctional non-catalytic component of the multisynthetase complex. The complex consists of nine catalytic and three non-catalytic proteins, which catalyze the ligation of amino acids to their cognate tRNA isoacceptors for use in protein translation. To date, two allelic variants in the AIMP1 gene have been reported as the underlying cause of autosomal recessive primary neurodegenerative disorder. Here, we present two consanguineous families from Pakistan and Iran, presenting with moderate to severe intellectual disability, global developmental delay, and speech impairment without neurodegeneration. By the combination of homozygosity mapping and next generation sequencing, we identified two homozygous missense variants, p.(Gly299Arg) and p.(Val176Gly), in the gene AIMP1 that co-segregated with the phenotype in the respective families. Molecular modeling of the variants revealed deleterious effects on the protein structure that are predicted to result in reduced AIMP1 function. Our findings indicate that the clinical spectrum for AIMP1 defects is broader than witnessed so far.
Similar content being viewed by others


Introduction
AIMP1/p43, which has both cytokine- and tRNA-binding activities, is a non-catalytic component of the mammalian multi-tRNA synthetase complex (MSC), which consists of nine different aminoacyl-tRNA synthetases (EPRS, IRS, LRS, MRS, QRS, RRS, KRS and DRS) and three auxiliary factors, AIMP1/p43, AIMP2/p38 and AIMP3/p18.1, 2, 3 The tRNA-binding domain associates with several aminoacyl-tRNA synthetases. On cleavage from the p43 component of the MSC, this domain becomes an independent domain with inflammatory cytokine activity. Mouse studies have shown neuronal expression of AIMP1 in a variety of regions of the central nervous system, including the ventral horn of the spinal cord and the hippocampus.4, 5
To date, two variants for the AIMP1 gene (OMIM *603605) have been reported and these are associated with severe neurodegenerative disorder. The first alteration is a homozygous 2-bp deletion (NM_001142415.1, c.292_293delCA, p.(Gln98Valfs*30)) in exon 4 of AIMP1 that was found in six affected individuals from three related families, as well as in one single individual of another very remotely related consanguineous family of the same tribe. The c.292_293delCA results in a frameshift and predicted truncation of the major conserved functional domains of the protein (tRNA-binding domain, a segment required for induction of inflammation, and regions that control angiogenesis).6 The second is a homozygous variant c.115C>T (NM_001142415.1) in exon 3, which introduces an early stop codon p.(Gln39*), identified in one affected female with Filipino ethnic origin.7 Major clinical features in patients with AIMP1 mutations, described so far, are listed in Supplementary Table S1. Affected individuals presented with a severe phenotype encompassing rapid and progressive neurological deterioration, severe failure to thrive and severe global developmental delay/intellectual disability (ID) with lack of speech, axial hypotonia, progressive spastic paraparesis and seizures.
ID is the most prevalent severe disability in children and affects 1–3% of the world's population.8, 9, 10, 11, 12 In affected children from consanguineous families, autosomal recessive (AR) inheritance is common;13 however, congenital disorders with ID constitute an important medical problem because they represent a challenge in diagnosis, high prevalence and life-long care that most of these patients need.14 A recent review suggests that ARID is not rare, and in outbred populations as many as 13–24% of ID may be due to AR genes,13 and identifying the underlying genetic cause is an important issue in clinical genetics.15
Consanguineous marriages, for which there is cultural preference in many countries including Iran, Pakistan and Syria, are an important risk factor for ARID and other congenital disorders,15 resulting in a significant excess of ID in offsprings of consanguineous marriages.14, 16, 17, 18
In the present study, we report on two consanguineous families with novel missense variants in AIMP1 that co-segregated with the ID phenotype. The first family, PKMR60 is from Pakistan and the affected family members present with moderate to severe ID. The second family, M105, is from Iran. Here, the affected individuals present with a history of developmental delay and moderate ID. For both novel allelic variants, we have performed in silico 3D modeling of the affected protein residues. The molecular modeling supported the pathogenic role of the identified alterations. The segregation data and overlapping clinical presentation of both families are also supportive for the pathogenic roles of the identified homozygous variants.
Materials and methods
Participants
Family PKMR60 is from central region of the province Punjab, Pakistan. The parents are consanguineous with 10 offspring and 5 of them are intellectually disabled (Figure 1a). This study was approved by the Institutional Review Board (IRB) of the Centre of Excellence in Molecular Biology (CEMB), the University of Punjab, Lahore, Pakistan and the local ethics committee of the Radboud University Medical Centre, Nijmegen, The Netherlands, and appropriate written informed consent was obtained from the parents.

(a) Pedigree structure of family PKMR60 and haplotype analysis. The inheritance pattern of nine markers on chromosome 4q is shown. The order of markers is depicted. The highlighted portion shows affected alleles in each individual. (b) Pedigree of Family M105 from Iran. Individuals V-1 and V-2 are dizygotic twins. Full symbols denote affected individuals with ID.
Iranian family (M105) is a five generations consanguineous family (Figure 1b), recruited by the Genetic Research Centre, Teheran, Iran. The consanguineous parents (father is deceased) had 6 children, 2 of them presented with moderate ID. Written informed consent was obtained from the mother. This study was performed in accordance with the ethical standards of the University of Social Welfare and Rehabilitation Sciences, Tehran, Iran. Genomic DNA was extracted from peripheral blood of affected and unaffected members of family PMKR60 and the patients and their mother of family M105 using standard procedures.19
Clinical evaluation
To rule out any other syndrome along with ID, the individuals (VI-1, VI-2, VI-3, VI-4 and VI-5) of family PKMR60 (two females and three males, aged 22, 19, 12, 14 and 21, respectively) were clinically evaluated. Biochemical tests and MRI scan were carried out on the index patient VI-1. Radiological and biochemical tests were carried out at Shaukat Khanum Memorial Cancer Hospital and Research Centre Lahore Pakistan.
The probands V-I and V-5 (male and female, aged 36 and 30, respectively) from family M105, were examined by experienced clinical geneticists who assessed their physical and mental status. To rule out chromosomal aberrations and fragile X-syndrome, karyotype analysis by G-banding, as well as fragile X testing by Southern blot analysis and PCR were carried out, respectively. The karyotype of all patients was normal and fragile X-syndrome was excluded.
Homozygosity mapping
For the detection of regions of homozygosity that the five affected individuals of PKMR60 have in common, their genomic DNAs were taken and mixed in equimolar concentrations. The mixture of five DNA samples was hybridized on the CytoScan HD array that contains over 2.6 Million probes of which ~750.000 are SNP probes (Affymetrix, Inc., Santa Clara, CA, USA). Hybridizations were performed according to the manufacturer’s protocol. SNPs and allele peak values were called using the Affymetrix Chromosome Analysis Suite (ChAS version 1.2.0.225) (Affymetrix, Inc.) with default references and settings. Data viewing and analysis was performed with ChAS software. Regions of homozygosity were considered to be in common in all five individuals when the average absolute allele peak value of the regions exceeded the value of 0.95.
For the detection of regions of homozygosity in common between the affected individuals in family M105, genomic DNAs from individuals V-1 and V-5 were mixed in equimolar concentrations. The DNA mixture was hybridized onto a Genome-wide Human SNP Array 6.0 (Affymetrix, Inc.). Data was analyzed with Partek Genomic Suite software (Partek Inc., St Louis, MO, USA).
Linkage analysis
Linkage analysis was performed by using fluorescently labeled microsatellite markers. The PCR products were pooled in deionized formamide containing the LIZ as size standard. The samples were denatured at 95 °C for 5 min. The amplified products were analyzed on the ABI Prism 3100 or 3730 DNA genetic analyzer (Applera Corp, Foster City, CA, USA) and alleles were assigned using GENESCAN, GENOTYPER and GENEMAPPER Software packages (Applied Biosystems, Foster City, CA, USA). All members of the family were genotyped and haplotypes were constructed to confirm the homozygosity and linkage.
Statistical evaluation of linkage
Two-point LOD scores were calculated for all markers within the homozygous interval by using Easy Linkage Software.20 We assumed a susceptibility allele with a frequency of 0.05 and a recessive mode of inheritance with complete penetrance. Recombination frequencies were assumed to be equal in both genders. Genetic distances were based on Marshfield human genetic map.21
Next generation sequencing
Whole exome sequencing—family PKMR60
The DNA of the index patient VI-1 was used for whole exome sequencing. Next generation sequencing and analysis was carried out as described before.22 In brief, exome enrichment was performed using the SureSelect Human All Exon 50 Mb Kit (Agilent, Santa Clara, CA, USA), covering ~21 000 genes. The enriched exome library was equimolarly pooled in a set of four samples including three other unrelated samples. The pool was based on a combined library concentration of 1 pM. Subsequently, the obtained pool was used for emulsion PCR and bead preparation using the EZbead system, following manufacturer's instructions (Life Technologies, Carlsbad, CA, USA). For each pool of four exome libraries, a full sequencing slide was used on a SOLiD 4 System (Life Technologies), thereby anticipating that all four samples would be represented as a quarter of the total beads sequenced.
Color space reads were mapped to the hg19 reference genome build with the SOLiD bioscope software v1.3 (Life Technologies). Exclusion of known variants was based on dbSNPv132 and an in-house database including in-house analyzed exomes. Thus, these filtering steps led to a list of private variants, as such, most likely to contain the alteration(s) affecting function. The next filtering step was based on a recessive model of disease, keeping those variants that occurred in >70% of all reads, suggestive for homozygous variants, and all genes with two (or more) variants (with variation between 20 and 80%), suggestive for compound heterozygosity. The variant data was submitted to Leiden Open Variant Database (LOVD, www.lovd.nl/AIMP1). The variant identification number is mentioned in Table 1.
Whole exome sequencing—family M105
Genomic DNA from index patient V-1 was used to generate Illumina PairEnd pre-capture library (SureSelect XT Target Enrichment System for Illumina Paired-End Sequencing Library; Agilent Technologies Inc., Wilmington, DE, USA) in accordance with the manufacturer's protocol (Agilent Technologies). The captured library was sequenced on HiSeq 2000 (Illumina) in accordance with the manufacturer's protocol, in a 101-nucleotide single-end sequencing format, with a coverage >97%.
Raw sequence reads were pre-screened to remove low-quality reads, and then aligned to the human reference genome (hg19, GRCh37) with SOAP (version 2.20; http://soap.genomics.org.cn/soapaligner.html/). Variant lists were filtered against dbSNPv137, whole genomes from 185 healthy individuals (1000 Genomes Project) and 200 exomes from Danish individuals, and 6500 exomes present in the Exome Variant Server (NHLBI GO Exome Sequencing Project, Seattle, WA (http://evs.gs.washington.edu/EVS/) 30 August 2013; v.0.0.21). In addition, variants were compared with in-house databases containing >200 exomes. Variants were ranked as potential candidates as previously described,23 using an improved version of Medical Resequencing Analysis Pipeline (MERAP) developed by Hu.24, 25 The OMIM catalog (http://www.ncbi.nlm.nih.gov/omim) and the Human Gene Mutation Database (HGMD, http://www.hgmd.org/) were used as a filter to identify all previously described affecting function variants. The variant data was submitted to LOVD, and variant identification number is mentioned in Table 1.
Sanger sequencing
For exclusion of technical artifacts and for conducting segregation analysis, the identified variants were confirmed by Sanger sequencing, which was performed according to standard protocols. Primers for the amplification of the change carrying exons were designed by using Primer3 software (http://bioinfo.ut.ee/primer3-0.4.0/).26 Sequence analysis was performed with the ABI PRISM Big Dye Terminator Cycle Sequencing V3.1 Ready Reaction Kit on the ABI PRISM 3730 DNA Analyzer or the 3500 Genetic Analyzer for Resequencing & Fragment Analysis (Applied Biosystems).
Results
Clinical description
Family PKMR60 was enrolled from Punjab, Pakistan and has five affected children and five unaffected siblings. The pedigree and facial features of the patients are shown in Figures 1a and 2, respectively. Results of the clinical evaluation of all affected members are shown in Table 1. All affected individuals present with moderate to severe ID without associated congenital malformations or facial dysmorphisms. Body height, weight and head circumference were normal. Hematological and biochemical parameters showed slight hematological abnormalities in patient VI-1. Both, the affected and the normal individuals (VI-1, VI-8) showed hyperglycemia (371 mg/dl and 342 mg/dl, respectively). Head MRI without contrast was performed on patient VI-1 and revealed no significant structural brain anomalies.

Facial features of affected individuals with mutations in the AIMP1 gene. VI-1 to VI-5 are patients from family PKRM60. The blue-rimmed picture shows the index patient V-1 from the Iranian family M105. A full color version of this figure is available at the European Journal of Human Genetics journal online.
In the consanguineous Iranian family M105 there are two affected offsprings (V-1 and V-5) presenting with moderate ID. Patient V-1 (Figure 2) is the first child of a non-identical twin pregnancy from the healthy consanguineous parents, originating from the central part of Iran. The patient was born at term after an uneventful pregnancy. Birth weight, length and OFC were not documented. He had global developmental delay and started to walk and spoke his first words at 4 years of age. No history of ataxia, muscular hypotonia or seizure was reported. His body height at age of examination (36 years) was 165 cm (−1 SD), weight was 64 kg (BMI: 23.5) and OFC was 55 cm (−1 SD). Facial appearance was non-dysmorphic and no physical malformations were observed. He made good eye contact, but showed very short temper and aggressive behavior. Expressive language was apparently better than receptive language associated with stutter during anxiety. Hearing was normal. Cognitive evaluation at 36 years of age (WAIS-IV) revealed an IQ of 54 in the range of moderate ID. The Achenbach Child Behavior Checklist (CBCL/6-18) assessed aggressive behavior.
Patient V-5 is the younger sister of patient V-1. She was born at term after an uneventful pregnancy. Birth weight, length and OFC were not documented. Global developmental delay was noted at 2 years of age, she started to walk and spoke her first words at 2.5 years of age. No seizures or ataxic gait were noted. At the age of examination (30 years), height was 153 cm (−1 SD), weight was 54 kg (BMI: 23.1) and head circumference was 53 cm (−1 SD). Cognitive evaluation at age 30 years (WAIS-IV) revealed an IQ of 41 in the range of moderate ID. She is taking care of her personal needs. She had impaired verbal expression and communication problems with stutter during anxiety. Visual testing showed myopia with no other abnormality. Facial appearance was normal.
Homozygosity mapping
Homozygosity mapping was performed by genotyping the pooled DNA samples of the five affected individuals (VI-1 to VI-5) of family PKMR60 using CytoScan HD array. Only a single large shared homozygous region was identified. The 22.2-Mb interval is located at chromosome 4q22.2–q26 between the SNP markers rs12642948 (chr4.hg19:g.94812609C>T) and rs13108295 (chr4.hg19:g.117055445T>C). Next, genotyping around this region by STR markers was performed for all healthy and affected family members to confirm the homozygosity of this region. A maximum two-point LOD score of 3.7 was obtained for markers D4S1572, D4S2309, D4S2297 and D4S2303 (Supplementary Table S2), providing strong evidence for an AR locus in family PKMR60. The proximal breakpoint between markers D4S2460 and D4S2986 confirmed the position of the breakpoint as observed by array analysis.
Homozygosity mapping was performed by genotyping pooled DNA samples of both affected individuals (V-1 and V-5) of family M105 using Generic Affymetrix Array and SNP 6.0 assay (Affymetrix, Inc.). Several shared homozygous intervals have been identified. Interestingly, the second largest shared region of homozygosity was a 32.9-Mb interval between SNPs rs6823261 (chr4.hg19:g.106478315A>G) and rs11731968 (chr4.hg19:g.139 405 009A>G) (Supplementary Table S3), which overlaps for ~10.5 Mb the common region of homozygosity in PKMR60 (chr4.hg19:g.106 478 315–117 055 445) (Figure 3).

A schematic diagram showing the shared homozygous region of 10.5 Mb (chr4:106 478 315–117 055 445) between the families PKMR60 and M105. The shared homozygous region is located at cytoband q24–q26. The figure is generated by using the ‘add custom tracks’ function of the UCSC genome browser (February 2009 (GRch37/hg19)).
Whole exome sequencing, validation and in silico analysis
The exome of patient VI-1 of family PKMR60 was sequenced using the 50 Mb SureSelect human exome kit, targeting 96.47% of all genes from the NCBI RNA reference collection. We obtained 3.72 Gb of mapable sequence with a mean coverage of 40-fold, with 78% of targets covering at least 5-fold. A total of 9173 variants were detected in the coding regions and canonical splice sites. We then applied a prioritization scheme to identify the pathogenic mutation, similar to the one described before.22 To this end, we excluded variants known in dbSNPv132, as well as variants from our in-house database. This analysis reduced the number of candidates to 540.
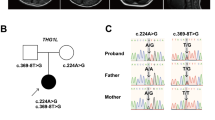
Based on a recessive inheritance model, applying a phyloP threshold >2.5 for non-synonymous variants22 and after inspecting the raw reads files, we identified 21 candidate genes: 19 genes with potential homozygous variants (determined by >70% variant reads) and 2 genes with potential compound heterozygous variants (>20–80% variant reads only a single gene, AIMP1 (OMIM:*603605) carrying a homozygous variant (NM_001142415.1, c.895G>A) was identified in the homozygous linkage region on chromosome 4q22.2–q26. This missense variant [chr4.hg19:g.107268805G>A, c.895G>A, p.(Gly299Arg)] was confirmed by Sanger sequencing and showed, as expected, complete segregation with the phenotype in family PKMR60 (Figure 4a). This specific variant was neither found in 262 ethnically matched chromosomes, in dbSNP v138, 1000 genomes data set nor in Exome Variant Server. The missense mutation had a high PhyloP score of 4.5 and a Grantham score of 125, and was predicted by Polyphen2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org/) to be damaging at the functional level. The amino acid Gly299 was found to be conserved down to Fruit fly, suggesting an important role in the normal function of the protein (Figure 5). Next, we have used project HOPE27 to perform 3D modeling of the identified missense change. The mutant residue (arginine) is larger in size than the wild-type residue. The wild-type amino acid is neutral, whereas the mutated amino acid is hydrophilic. So, the mutant residue introduces a charge in a buried residue (in the core of the protein), which can result in protein-folding problems. The mutant residue is bigger and will introduce inappropriate torsion angles. Only glycine is flexible enough to make these torsion angles; mutation into another residue will force the local backbone into an incorrect conformation and will probably disturb the local structure (Figures 6a and b).

(a) Sequence chromatograms showing the complete segregation of the missense mutation c.895G>A in family PKMR60 in the patients (left), the healthy sibling (middle) and both parents (right). Only one healthy sibling (Vl-6) is homozygous for the wild-type allele. (b) Sequence chromatograms of both affected members and the mother of family M105 show c.527T>G in AIMP1 in a heterozygous (IV-2) and homozygous (V-1 and V-5) state compared with the wild-type sequence. A full color version of this figure is available at the European Journal of Human Genetics journal online.

Partial amino acid sequence alignment of the human AIMP1 protein and its homologs in the common model systems. The positions of the missense mutations p.(Val176Gly) family M105 and p.(Gly299Arg) family PKRM60 are indicated by an arrow.

(a) Family PMKR60: ribbon presentation of the AIMP1 protein. The protein is colored gray, the side chain of the mutated residue, p.(Gly299Arg), is colored magenta and shown as small balls. (b) Close-up of the p.(Gly299Arg) mutation. The protein is colored gray, the side chains of both the wild-type and the mutant residue are shown and colored green and red, respectively. (c) Family M105: overview of the protein in ribbon presentation, indicating the position of the p.(Val176Gly) substitution (magenta and ball representation) (d) Close-up of the p.(Val176Gly) mutation, showing the side chain of the wild-type (green) and the mutant (red) residue. A full color version of this figure is available at the European Journal of Human Genetics journal online.
The exome of patient V-1 from family M105 was sequenced using SureSelect human exome kit, targeting the coding sequences of RefSeq genes (hg19; 19 083 genes, 33.3 Mb). The sequence analysis was conducted by an in-house pipeline (https://sourceforge.net/projects/merap). Non-synonymous and splice-site variants matching the recessive model and located in the homozygous shared intervals were retained, and filtered against the polymorphism databases (dbSNPv137, 1000Genome, exome variant server and an in-house exome database of 417 individuals). The analysis of prospective changes indicated DNA variants in three genes: AIMP1 [OMIM:*603605; chr4.hg19:g.107252964T>G, NM_001142415.1 c.527T>G, p.(Val176Gly)], ARHGEF38 [chr4.hg19:g.106588622C>T, NM_001242729.1 c.1910C>T, p.(Ser637Phe)] and CASKIN1 [OMIM: *612184; chr16.hg19: g.2237169C>T, NM_020764 c.833G>A, p.(Arg278Gln)]. The first two changes were located in a 33 Mb shared homozygous region on 4q24–q28.3, between SNP markers rs6823261 and rs11731968, whereas the third variant was identified in a ∼5 Mb homozygous region on 16p13.3, between SNP markers rs41340949 (chr16.hg19:g.86671A>G) and rs8060791 (chr16.hg19:5231188A>C). The co-segregation pattern of the three variants was checked in the family, and all variants segregated correctly including the AIMP1 variant (Figure 4b). By using different pathogenicity evaluators (Polyphen2, SIFT), the AIMP1 mutation was ranked as the most plausible pathogenic change in family M105.
The AIMP1 c.527T>G mutation was found to be absent from 476 chromosomes from ethnically matched Iranian controls, exome variant server, as well as dbSNPv138. The amino acid residue affected by the mutation p.(Val176Gly) is highly conserved from human to fruit fly (Figure 5). The 3D modeling of the p.(Val176Gly) change revealed that the introduced glycine is a very flexible residue and can disturb the required rigidity of AIMP1 at this position. In addition, the mutant residue is smaller compared with the wild-type residue and the change causes loss of hydrophobic interaction in the protein core. Close-up views of the wild-type and mutant protein positions are shown in Figures 6c and d. Furthermore, based on Uniprot analysis, Project HOPE revealed that p.(Val176Gly) is located in an area of the protein, which is part of two different protein domains. The first domain is responsible for tRNA binding and the second domain, designated HSP90B1, is involved in endothelial cell migration.
Discussion
In this study we present two novel missense variants in AIMP1, that are associated with moderate to severe ID, and global developmental delay in Pakistani and Iranian families. The observed ID phenotypes are relatively mild in comparison to previous reported patients with AIMP1 mutations, who presented a considerable more severe phenotype including neurologic deterioration.6, 7 Thus our data show a broadening of the phenotypic spectrum associated with AIMP1 allelic variants.
A homozygous frameshift and a stop codon change in AIMP1 have been associated with severe primary neurodegenerative disorder. In the first case, the variant is predicted to lead to a truncated AIMP1 protein lacking the main functional domains, including the tRNA-binding domain.6 In the second family, the early premature stop codon very likely leads to degradation of the AIMP1 transcript by non-sense-mediated mRNA decay and therefore no production of functional AIMP1 protein is expected.7 The phenotype observed in these patients corresponds well to the findings observed in mice lacking the AIMP1/p43 orthologue.5 Furthermore, it has been shown that axon integrity in the central and peripheral neural system could not be maintained in these animals. In addition, expression studies of mouse Aimp1 have provided evidence that this gene is expressed in neurons in a variety of regions of the central nervous system, including the hippocampus and the ventral horn of the spinal cord.5
The severe neurologic features of the first reported patients with a frameshift mutation in AIMP1 led to the clinical classification of a Pelizaeus–Merzbacher-like phenotype.6 However, this designation has been debated and currently ‘primary neurodegenerative disease’ is the favored classification.6, 7, 28, 29 The patients with truncating mutations in AIMP1 present with a severe phenotype encompassing a progressive neurological deterioration, severe failure to thrive and severe global developmental delay/ID, axial hypotonia, progressive spastic paraparesis, and seizures. Furthermore, their MRI scans show arrest of myelination. In addition, magnetic resonance spectroscopy demonstrated a relative decrease of N-acetylaspartate within the white matter as a result of axonal degeneration and generalized brain atrophy, especially of the corpus callosum, was observed.6, 7 In particular, in the patient described by Armstrong et al,7 subsequent MRIs (within a 2-month interval) revealed myelin deficiency and progressive cerebral atrophy, consistent with a primary neuronal degenerative disorder with a rapid clinical course leading to a premature death. The authors of the latter mentioned study proposed that AIMP1 deficiency should be added to the differential diagnosis of infantile onset, progressive neurodegenerative disease.7 AIMP1 interacts with a subtype of neurofilament (light subunit, NF-L), which forms part of the cytoskeleton that confers the intracellular scaffold and mechanical stability of neurons, and furthermore regulates their assembly by altering their phosphorylation level.5 Neurofilaments are dynamic structures known to play roles in neuronal development and function, such as neuronal morphogenesis, migration, axonal growth, synaptic plasticity and alteration of their phosphorylation state has been involved in the onset of neurodegenerative disorders,30, 31 suggesting that the impaired myelin formation in the patients with truncating AIMP1 variants is secondary to neuronal dysfunction.
The affected individuals of both families presented here, show a mild phenotype as compared with the severe primary neurodegenerative disorder patients. We have performed MRI for the index patient (VI-1) of the Pakistani family, PKMR60. No hypomyelination or axonal degeneration in the white matter of the brain could be observed and no intra- or extra-axial masses were seen. Furthermore, we noted normal ventricular system, seventh and eighth nerve complexes, posterior fossa and temporal lobe. Most importantly, the myelination pattern also appeared to be normal. In addition, hyperglycemic features were observed in both affected and unaffected individuals of family PKMR60, suggesting that hyperglycemia cannot be attributed directly to the identified AIMP1 variant in this family. Also the absence of hyperglycemia in other patients with an AIMP1 mutation suggests that this feature is likely to be caused by another factor.
The mild phenotype of the affected individuals of the Pakistani (PKMR60) and Iranian (M105) families shows overlapping features including the category of ID (moderate to severe), delayed global development and delayed speech acquisition, aggressiveness, normal walking and gait, no ataxia or seizures and no progressive course of disease. The difference in the phenotypic presentations compared with primary neurodegenerative disorder patients might be explained by the different types of changes in the AIMP1 gene and their effect on protein function. While the alterations underlying neurodegenerative disorder are truncating variants, most probably resulting in lack of AIMP1, we observed two distinct missense variants that alter evolutionary highly conserved residues of the AIMP1 protein.
The first change, (c.895G>A, p.(Gly299Arg)); family PKMR60, is located in the COOH-terminal domain and results in the insertion of an amino acid residue that is larger in size as compared with the wild-type residue and might introduce inappropriate torsion angles that can disturb the local structure. Furthermore, arginine is hydrophilic and introduces a charge in a buried residue of the protein, which could result in protein-folding problems.
The second missense variant (c.527T>G, p.(Val176Gly; family M105)) affect a highly conserved amino acid, and the mutant residue (glycine) is smaller in size than the wild-type residue. In addition, the amino acid change causes loss of hydrophilic interactions in the protein core, and thus could affect the core structure. Furthermore, altered amino acid is located in a domain responsible for tRNA binding, which could disturb or even abolish the domain’s function. Both novel variants affect crucial domains of the AIMP1 protein and could lead to severe deleterious effects by introducing conformational changes or disturbing tRNA binding, respectively. Thus, both amino acid changes can disrupt the function of the whole MSC, leading to profound consequences for cell metabolism.
In conclusion, our study broadened the phenotypic spectrum of AIMP1 defects. There is growing evidence that variants affecting function, in the currently known genes involved in syndromic and non-syndromic forms of ID, are only the tip of the iceberg. Identification of additional variants and understanding their impact on gene function will not only improve genetic counseling of affected families, but will furthermore advance understanding of molecular networks involved in cognitive processes, which may result in studying treatment and therapy of cognitive impairment in the future.15, 32
References
Quevillon S, Agou F, Robinson JC, Mirande M : The p43 component of the mammalian multi-synthetase complex is likely to be the precursor of the endothelial monocyte-activating polypeptide II cytokine. J Biol Chem 1997; 272: 32573–32579.
Quevillon S, Mirande M : The p18 component of the multisynthetase complex shares a protein motif with the beta and gamma subunits of eukaryotic elongation factor 1. FEBS Lett 1996; 395: 63–67.
Quevillon S, Robinson JC, Berthonneau E, Siatecka M, Mirande M : Macromolecular assemblage of aminoacyl-tRNA synthetases: identification of protein-protein interactions and characterization of a core protein. J Mol Biol 1999; 285: 183–195.
Lee SW, Cho BH, Park SG, Kim S : Aminoacyl-tRNA synthetase complexes: beyond translation. J Cell Sci 2004; 117: 3725–3734.
Zhu X, Liu Y, Yin Y et al: MSC p43 required for axonal development in motor neurons. Proc Natl Acad Sci USA 2009; 106: 15944–15949.
Feinstein M, Markus B, Noyman I et al: Pelizaeus-Merzbacher-like disease caused by AIMP1/p43 homozygous mutation. Am J Hum Genet 2010; 87: 820–828.
Armstrong L, Biancheri R, Shyr C et al: AIMP1 deficiency presents as a cortical neurodegenerative disease with infantile onset. Neurogenetics 2014; 15: 157–159.
Hamdan FF, Gauthier J, Spiegelman D et al: Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N Engl J Med 2009; 360: 599–605.
Chelly J, Khelfaoui M, Francis F, Cherif B, Bienvenu T : Genetics and pathophysiology of mental retardation. Eur J Hum Genet 2006; 14: 701–713.
Garshasbi M, Hadavi V, Habibi H et al: A defect in the TUSC3 gene is associated with autosomal recessive mental retardation. Am J Hum Genet 2008; 82: 1158–1164.
Kuss AW, Garshasbi M, Kahrizi K et al: Autosomal recessive mental retardation: homozygosity mapping identifies 27 single linkage intervals, at least 14 novel loci and several mutation hotspots. Hum genet 2010; 129: 141–148.
Leonard H, Wen X : The epidemiology of mental retardation: challenges and opportunities in the new millennium. Ment Retard Dev Disabil Res Rev 2002; 8: 117–134.
Musante L, Ropers HH : Genetics of recessive cognitive disorders. Trends Genet 2014; 30: 32–39.
Iqbal Z, Neveling K, Razzaq A et al: Targeted next generation sequencing reveals a novel intragenic deletion of the TPO gene in a family with intellectual disability. Arch Med Res 2012; 43: 312–316.
Iqbal Z, van Bokhoven H : Identifying genes responsible for intellectual disability in consanguineous families. Hum Hered 2014; 77: 150–160.
Bittles A : Consanguinity and its relevance to clinical genetics. Clin Genet 2001; 60: 89–98.
Al-Ansari A : Etiology of mild mental retardation among Bahraini children: a community-based case control study. Ment Retard 1993; 31: 140–143.
Bundey S, Alam H, Kaur A, Mir S, Lancashire R : Why do UK born Pakistani babies have high perinatal and neonatal mortality rates? Paediatr Perinat Epidemiol 1991; 5: 101–114.
Grimberg J, Nawoschik S, Belluscio L, McKee R, Turck A, Eisenberg A : A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res 1989; 17: 8390.
Lindner TH, Hoffmann K : easyLINKAGE: a PERL script for easy and automated two-/multi-point linkage analyses. Bioinformatics 2005; 21: 405–407.
Broman KW, Murray JC, Sheffield VC, White RL, Weber JL : Comprehensive human genetic maps: individual and sex-specific variation in recombination. Am J Hum Genet 1998; 63: 861–869.
Vissers LE, de Ligt J, Gilissen C et al: A de novo paradigm for mental retardation. Nat Genet 2010; 42: 1109–1112.
Najmabadi H, Hu H, Garshasbi M et al: Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 2011; 478: 57–63.
Reuter MS, Musante L, Hu H et al: NDST1 missense mutations in autosomal recessive intellectual disability. Am J Med Genet A 2014; 164A: 2753–2763.
Hu H, Wienker TF, Musante L et al: Integrated sequence analysis pipeline provides one-stop solution for identifying disease-causing mutations. Hum Mutat 2014; 35: 1427–1435.
Rozen S, Skaletsky H : Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 2000; 132: 365–386.
Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G : Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics 2010; 11: 548.
Biancheri R, Rossi A, Zara F, Filocamo M : AIMP1/p43 mutation and PMLD. Am J Hum Genet 2011; 88: 391, author reply 393-395.
Boespflug-Tanguy O, Aubourg P, Dorboz I et al: Neurodegenerative disorder related to AIMP1/p43 mutation is not a PMLD. Am J Hum Genet 2011; 88: 392–393, author reply 393-395.
Laser-Azogui A, Kornreich M, Malka-Gibor E, Beck R : Neurofilament assembly and function during neuronal development. Curr Opin Cell Biol 2015; 32C: 92–101.
Lepinoux-Chambaud C, Eyer J : Review on intermediate filaments of the nervous system and their pathological alterations. Histochem Cell Biol 2013; 140: 13–22.
Abou Jamra R, Wohlfart S, Zweier M et al: Homozygosity mapping in 64 Syrian consanguineous families with non-specific intellectual disability reveals 11 novel loci and high heterogeneity. Eur J Hum Genet 2011; 19: 1161–1166.
Acknowledgements
We wish to thank the members of the families PKMR60 and M105 who voluntarily participated in this study. We thank Bettina Lipkowitz, Melanie Bienek, Sabine Otto and Vanessa Suckow for expert technical assistance. This research has received funding from the European Union’s Seventh Framework Program under grant agreement number 241995 (project GENCODYS). H-HR was funded by the Max-Planck Society, and AR, MYZ and ZI were supported by the Higher Education Commission of Pakistan (HEC).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Iqbal, Z., Püttmann, L., Musante, L. et al. Missense variants in AIMP1 gene are implicated in autosomal recessive intellectual disability without neurodegeneration. Eur J Hum Genet 24, 392–399 (2016). https://doi.org/10.1038/ejhg.2015.148
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.148
This article is cited by
-
The recurrent missense mutation p.(Arg367Trp) in YARS1 causes a distinct neurodevelopmental phenotype
Journal of Molecular Medicine (2021)
-
Roles of aminoacyl-tRNA synthetase-interacting multi-functional proteins in physiology and cancer
Cell Death & Disease (2020)
-
Aminoacyl-tRNA synthetases as therapeutic targets
Nature Reviews Drug Discovery (2019)
-
Pathogenic variants in AIMP1 cause pontocerebellar hypoplasia
neurogenetics (2019)
