
Abstract
Baraitser–Winter, Fryns–Aftimos and cerebrofrontofacial syndrome types 1 and 3 have recently been associated with heterozygous gain-of-function mutations in one of the two ubiquitous cytoplasmic actin-encoding genes ACTB and ACTG1 that encode β- and γ-actins. We present detailed phenotypic descriptions and neuroimaging on 36 patients analyzed by our group and six cases from the literature with a molecularly proven actinopathy (9 ACTG1 and 33 ACTB). The major clinical anomalies are striking dysmorphic facial features with hypertelorism, broad nose with large tip and prominent root, congenital non-myopathic ptosis, ridged metopic suture and arched eyebrows. Iris or retinal coloboma is present in many cases, as is sensorineural deafness. Cleft lip and palate, hallux duplex, congenital heart defects and renal tract anomalies are seen in some cases. Microcephaly may develop with time. Nearly all patients with ACTG1 mutations, and around 60% of those with ACTB mutations have some degree of pachygyria with anteroposterior severity gradient, rarely lissencephaly or neuronal heterotopia. Reduction of shoulder girdle muscle bulk and progressive joint stiffness is common. Early muscular involvement, occasionally with congenital arthrogryposis, may be present. Progressive, severe dystonia was seen in one family. Intellectual disability and epilepsy are variable in severity and largely correlate with CNS anomalies. One patient developed acute lymphocytic leukemia, and another a cutaneous lymphoma, indicating that actinopathies may be cancer-predisposing disorders. Considering the multifaceted role of actins in cell physiology, we hypothesize that some clinical manifestations may be partially mutation specific. Baraitser–Winter cerebrofrontofacial syndrome is our suggested designation for this clinical entity.
Similar content being viewed by others


Introduction
Background
Baraitser–Winter malformation syndrome (BWMS), characterized by short stature, hypertelorism, bilateral ptosis, ocular colobomata, metopic ridging and agyria/pachygyria, was delineated in 1988.1 Three patients were described in the original paper.1 The two siblings reported in this article probably did not have the BWMS, based on our current knowledge, but patient 3 (patient A2) had the typical facial features and pachygyria was subsequently reported in this child by Jeanette Ramer, who reported several other patients2, 3 (patients B3, B17 and A3). She emphasized the metopic ridging and included cerebral anomalies in the definition of BWMS. An apparently distinct entity was reported in 2000 in two patients with ptosis, a large nose, webbed neck, frontal pachygyria and limited joint extension,4 defined as Fryns–Aftimos syndrome (FA). On the basis of a review of several case reports, Robin Winter delineated three craniofrontofacial syndromes (CFF) for a series of patients with frontonasal dysmorphism, dysplastic ears and central nervous system (CNS) anomalies.5 CFF1 was associated with periventricular nodular heterotopia and agenesis of the corpus callosum. CFF2 had dilated Virchow–Robin spaces, but no migration defects. CFF3 was defined by the absence of heterotopia and of abnormal Virchow–Robin spaces, but presence of pachygyria. Winter noted: ‘there does seem to be considerable overlap between the three groups [of CFF] suggesting a common embryological pathway, if not allelic mutations’. CFF3 was a heterogeneous group, encompassing several reports of patients with macroblepharon, eyelid coloboma, ear anomalies, macrostomia, coronal craniosynostosis, cortical atrophy and/or agenesis of the corpus callosum, amongst which Winter included FA cases. Recently, BWMS, FA and some patients with CFF3 were associated with missense mutations in one of the two ubiquitous cytoplasmic β- and γ-actin-encoding genes ACTB (7p22.1) and ACTG1 (17q25.3)6, 7, 8, 9
In this article, we discuss 42 patients with mutations in these genes with BWMS, FA or CFF. Having identified identical mutations in patients carrying a clinical diagnosis of BWMS, FA or CFF, we propose a unified designation: Baraitser–Winter cerebrofrontofacial syndrome (BWCFF).
Patients recruitment and inclusion criteria
Our study has been carried out in collaboration with clinicians from different countries. Some patients were reported before the discovery of the genes (see Table 1). Mutation analysis was performed using Sanger sequencing in four laboratories (initial research in Seattle and Nijmegen, routine in Paris and Dresden). We gathered 42 patients, including the 16 patients briefly reported in our first paper,7 four newly published patients with mutations6, 8, 9 and twins with an ACTB mutation, who developed dystonia in late childhood and died in their early twenties,10, 11 as, retrospectively, the diagnosis in these patients was compatible with BWCFF (B26 and B27).
We have also identified several patients with BWCFF-like phenotypes, in whom ACTB and ACTG1 screening was negative. Considering the wide spectrum of features observed in these BWCFF-like patients, and the lack of objective minimal criteria, we opted not to discuss this group.
Results
We identified 33 patients with ACTB and nine patients with ACTG1 mutations (Table 1 and Figure 1). The clinical data are summarized in Table 2. Figures 2, 3, 4 illustrate the dysmorphology and Figure 5 the neuroimaging. Unpublished mutations have been submitted to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/).

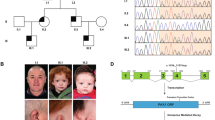
Mutation spectrum of BWCFF syndrome. The scheme takes advantage of the similarity of exon organization and protein sequence. Non-coding exon sequences are narrower and lighter. Functional domains are coded in color (apparent dispersion of each domain explained by 3D configuration). The number of the first aminoacid of each exon, and of the last aminoacid of exon 5 are mentioned. The full colour version of this figure is available at European Journal of Human Genetics online.

The spectrum of facial dysmorphology of BWCFF. Facial appearance of 15 patients with ACTG1 (An) or ACTB (Bn) mutations, subjectively sorted by gradient of severity. Picture of Patient B32 was previously published.41

Evolution of the phenotype with time. Evolution of the facial appearance of five patients with ACTG1 (An) or ACTB (Bn) mutations.

Non-facial anomalies in BWCFF. Patient B34: preaxial polydactyly of feet – Patient B6: pectus excavatum – Patient B19: brachydactyly and nail hypoplasia – Patient B11: Noonan-like appearance of mild BWCFF syndrome – Patient B32: severe CFF appearance: note bifid nose, narrow shoulder girdle with hypoplastic muscles webbed neck and severe scoliosis – Patient A1: note typical stance of older BWCFF patients, with anteverted shoulders and semiflexed knees.

MRI aspects of BWCFF – ACTB and ACTG1. T1 and T2-weighted axial and T1-weighted sagittal MR images ordered by extent of the cortical malformation. ACTG1: focal (central) pachygyria (A6), pachygyria and posterior subcortical band heterotopia (A2: white arrow) and diffuse pachygyria (A8, A3). Patient B22 also had a number of periventricular nodular heterotopia (see8). ACTB: normal cortex (B12), focal (central) pachygyria (B2 B22; white arrows), predominantly frontoparietal pachygyria (B9, B5), diffuse pachygyria (B8, B14), lissencephaly (B3). Note the shortened and thick corpus callosum (B2, B5, B14 and A8).
Craniofacial anomalies
Facial dysmorphism varies from mild to severe (Figures 2 and 3), which explains why actinopathies were previously divided into separate entities. The face is round and flat in infancy. This appearance may be enhanced by metopic ridging or trigonocephaly (26/40–65%), in association with relative hypoplasia of the malar regions. The median part of the face is broad with a wide nasal base and marked hypertelorism (39/41–95%). In severe cases, the face is reminiscent of frontonasal dysplasia (the ‘CFF variant’). With age there is an increasing fullness of the facial features, which contributes to the coarser appearance of BWCFF patients in adulthood (the ‘FA variant’).
The nose is wide, short and upturned with a large, flat tip (35/41–85%), a thick columella and everted, thick nares. A median grove may be present in very severe cases (Figure 2, A7, B32). In profile, the nose is short, and the dorsum is often in line with the forehead. With time, the nasal root becomes even more prominent, but remains flat in its middle part. The nose itself increases in thickness. The philtrum is long and smooth (32/38–84%) and ends in a thin vermillion border. There is retrognathia, a pointed chin, a wide mouth with downturned corners and an everted lower lip, which may be secondary to retrognathia (17/39–44%). The palate is highly arched. Four patients had cleft lip and palate. The facial appearance evolves considerably over time (Figure 3).
Bilateral ptosis is almost universal (37/40–93%) and may require surgery. Patients have long, often downslanting palpebral fissures, with or without epicanthus or epicanthus inversus, which may resemble the ocular appearance in Kabuki syndrome. The eyebrows are usually arched (35/40–88%) and are in continuity with the lateral edges of the nose; this appearance increases with age, and is, in part, the result of trying to raise upper eyelids. Uni- or bilateral ocular colobomata are common (11/40–28%) and may extend from the iris to the macula. Microphthalmia is present in a minority of cases (3/31–10%) and is not related to the extent of the colobomatous fissure. One of the male twins reported by Gearing et al10 developed early onset cataracts, not present in his twin brother. Coloboma is not obviously correlated with CNS malformations.
The ears are often small, posteriorly rotated, sometimes anteverted, with an overfolded, thick helix and a poorly developed antihelix (30/41–73%). They tend to be C or triangular shaped. Some patients have a hearing deficit of variable degree (13/40–33%), which can be progressive. Interestingly, deafness is observed with gain-of-function mutations in both ACTB and ACTG1, whereas isolated deafness has only been reported with loss-of-function mutations of ACTG1 (DFNA 20/26-OMIM 604717).
BWCFF shows variable muscular involvement, which confirms the involvement of ‘non-muscular’ actins in the development and maintenance of muscle. Reduction of the upper limb girdle muscle bulk variably affects many patients, and contributes to the appearance of narrow, sloping shoulders (Figure 4). This aspect is striking in patients B32 and B22, who had congenital arthrogryposis. Axillary and popliteal pterygia may be present. Many patients have a peculiar stance with dorsal kyphosis, anteverted shoulders and slightly flexed elbows and knees (Figure 4, A1), which may be associated with limited joint movements, and appears to progressively worsen in some. Ambulation becomes difficult is some adults, and may indicate a slowly progressive myopathic process. Camptodactyly and talipes are reported in patients with severe neurological impairment. Scoliosis is uncommon. The dystonia observed in the twin patients B26 and B27 may be unique. Both also had progressive esophageal achalasia presenting at the age of 2 years, which was surgically corrected in one. No smooth muscle-related problems have been reported in other patients with BWCFF.
CNS
Psychomotor development in BWCFF is highly variable. In children with normal brain architecture, there was motor delay, but otherwise development was only mildly to moderately delayed. Among those with a neuronal migration defect, development is always delayed, from profound ID in the patient with lissencephaly to mild ID in some patients with anterior pachygyria.
The spectrum of CNS anomalies is variable (Figure 5). Structurally, patients with BWCFF may have a normal brain or a migration disorder that, in most patients, presents with an anterior to posterior gradient of severity. When present, anomalies range from frontal pachygyria, predominantly central pachygyria to (rarely) lissencephaly. Subcortical band heteropia was seen in some (6/31–20%). A few periventricular heterotopia may be present. The corpus callosum often appears short and thick. Agenesis of the corpus callosum was seen with or without a cortical malformation in nine patients. The white matter is normal or, rarely, shows delayed myelination. The lateral ventricles may be mildly enlarged. Infratentorial anomalies are not present. Pachygyria/lissencephaly is present in at least 67% and appears more consistent in those with an ACTG1 mutation (Table 1). No cortical abnormality was reported in 13 patients on neuroimaging (of variable quality) or post mortem (B26 and B27). Head circumference tends to be low normal at birth. Mild postnatal microcephaly is present in about 50%. Spasticity was seen in severe cases.
Epilepsy was common (20/38–53%) but not consistent in patients with pachygyria or other structural cerebral anomalies. This may be related to the age of onset of seizures, which ranged from 2 months to 14 years. The epilepsy was reported as intractable in some individuals. BWCFF patients with a normal MRI scan did not have a seizure disorder. Sensorineural deafness was reported in a significant number of individuals. Only a few of them only had imaging of the inner ear, which demonstrated cochlear malformations in one case.
Neuropathologic investigations have only been performed in patients B26 and B27, who died in their early 20 s, and, considering their specific neurological history, we cannot draw general conclusions from their findings. These twins presented with mild developmental delay and developed sensorineural deafness at the age of 4 years. Onset of DOPA-unresponsive dystonia was around 12 years, followed by progressive cognitive decline. The brains were macroscopically unremarkable. Histology showed unusual fibrillary astrocytes in multiple regions and moderate iron accumulation in pallidal and nigral neurons. The most striking feature was extensive aggregation of actin and/or the actin-associated protein actin depolymerizing factor in eosinophilic rod-like structures in neurons of the neocortex and basal ganglia. This neurodegeneration may represent a neuropathologic evolution specifically linked to the p.Arg183Trp mutation. Alternatively, this mutation may be associated with an accelerated degenerative process that could affect other patients at a lower rate. Although some of our patients have lost to a degree their ability to walk, they have not manifested dystonic changes.
Growth and additional malformations
Intrauterine growth is usually normal in BWCFF. Moderate short stature is observed in teenagers and adults. This can be exaggerated by orthopedic problems. Patients with a severe phenotype tend to be shorter. Occasional malformations in BWCFF include pectus deformities, which contribute to their resemblance to Noonan syndrome. Congenital heart defects (CHD) were present in 12/36 (33%), and included bicuspid aortic valve, mitral valve regurgitation, ventricular septal defect, atrial septal defect and persistent ductus arteriosus; however, no clear CHD pattern emerged. Hydronephrosis (3 patients), horseshoe or ectopic kidneys and renal duplication (four patients), were observed in patients with ACTB mutations. Broad thumbs were sometimes mentioned, and duplication of the hallux was present in three CFF3 patients with ACTG mutation.
We have few data on prenatal findings. Polyhydramnios and nuchal translucency were reported in some cases.
Malignancies
A cutaneous lymphoma was diagnosed at the age of 19 years in patient A1 (61458 in7 and patient 3 in8), with a p.Thr120Ile mutation in ACTG1. Patient B25, who carries a p.Val209Leu mutation in ACTB, developed precursor B-cell acute lymphatic leukemia (ALL) at the age of 8 years and was treated according to the Dutch Childhood Oncology Group ALL-9 high-risk group protocol. He achieved complete remission and is healthy at the age of 21 years.
Discussion
Actins
Protein polymers forming the cytoskeleton include microtubules, actin filaments and intermediate filaments. Actin filaments provide mechanical intracytoplasmic supports, tracks for movements of intracellular components and forces to drive cell movements. They form relatively stable polymers (F-actins or stress fibers) that interact with myosin motor proteins to produce a sliding process at the origin of cytoplasmic movements involved in cell migration, cell shaping, cell division and muscle contraction (myofibrils). Actins may also show fast polymerizing/depolymerizing transition, necessary for adaptive cytoskeleton plasticity. Hundreds of accessory proteins are necessary to maintain the actin monomer pools and regulate actin filaments.12, 13
The actin gene family consists of six different isoforms with a sequence similarity >93%. Four actins are muscle specific (αsmooth and γsmooth actins in smooth muscle, αcardiac actin in cardiac and αskeletal actin in striated muscles). Only βcyto and γcyto actins are ubiquitously expressed. They have very similar protein sequences (Figure 1). They differ by only four biochemically similar amino acids within the 10N-terminal residues.14, 15 In contrast, their cDNA sequences differ by more than 11%, resulting in distinct 3D mRNA configurations.
ACTB and ACTG1 are highly expressed in non-muscular tissues and undifferentiated myoblasts, and minimally in differentiated myocytes.16, 17 The two proteins differ significantly by their subcellular localization, as was shown in myoblasts18 and neurons.19 They also have different post-translational modifications, for instance arginylation of β-actin, but not of γ-actin.20 ACTB and ACTG1 can copolymerize easily in vitro14 and are likely to act similarly in vivo, at least in lamellipodia.21 Consequently, ACTB and ACTG1 have both cooperative interactions and partially non-redundant functions.15, 22, 23 In muscle tissue, ACTG has been shown to have a role in sarcomere assembly.24 Tissue-specific inactivation of Actg in mice does not interfere with embryonic development of the muscles, but mice show muscle weakness with a progressive pattern of muscle fiber necrosis and regeneration, indicating that Actg must have a crucial function in the adult skeletal muscle. In the muscle cell lineage, ACTB expression is downregulated during muscle maturation, in contrast to ACTG1, which remains localized in the sarcolemma of mature skeletal muscle.25 Conditional muscle KO for Actg1 results in a progressive centronuclear myopathy.26
ACTB is deregulated in multiple malignancies including hepatocellular carcinoma, melanoma, ovarian cancers leukemia and lymphoma. The abnormal expression and polymerization of ACTB and the resulting changes to the cytoskeleton were shown to be associated with the invasiveness and metastasizing of these cancers.27 Recurrent somatic ACTB mutations were found in 5/55 diffuse large B-cell lymphomas.28 This finding is compatible with previous observations, that the cytoskeleton has a pivotal role in regulating the early events of B-cell activation.29 Actin and actin-related proteins are subunits of the BAF/PBAF (Polybromo-associated- BRG1- or HRBM-associated factors) chromatin remodeling complex (homolog to the SWI/SNF or SWItch/Sucrose Non-Fermentable complex of yeast), which regulates DNA transcription, synthesis and repair. Inactivating mutations in catalytic and regulatory subunits of this chromatin remodeling complex have been identified in human cancers.30, 31 We hypothesize that some actin mutations may be oncogenic by interfering with chromatin remodeling regulation. We screened a cohort of 95 B-cell ALL samples: no somatic ACTB mutations were identified, indicating that ACTB has at most a marginal role in sporadic hematologic carcinogenesis.
Genotype/phenotype correlations
Amino acid changes in ACTB and ACTG1 similarly result in BWCFF, confirming their functional complementarity in developmental pathways. Amino acid 196 in ACTB is a hotspot with 14 affected patients in our cohort: eight with an ACTB p.Arg196His and six with p.Arg196Cys mutation. Clinical heterogeneity among these patients with an identical molecular defect demonstrate intrinsic phenotypic variation of BWCFF.
If the phenotype is qualitatively identical with both genes, our series suggests that ACTG1 mutations may have a more central role in neuronal migration, as 8/9 ACTG1 patients have a migration disorder versus one-third of those carrying ACTB mutations (Table 2), although this difference does not reach statistical significance due to the small number of ACTG1 cases. Two patients reported by Di Donato have an analogous p.Thr120Ile mutation in ACTB and ACGT1. Considering that the patient with ACTB variant was much more affected, and that β-actin-knockout mice die during early embryonic development whereas ϒ-actin-knockout mice survive, she suggested that ACTB mutations may be associated with a more severe phenotype.8 Indeed, the three most severe cases in our series (B22, B32 and B34) carry the same p.Thr120Ile mutation. This may point even to a more subtle, mutation based genotype/phenotype correlation.
The gain-of-function mechanism of ACT mutations in BWCFF also implies that there could be an important mutation-specific variability in the phenotype of actinopathies. The morphology and the F-actin organization in lymphoblastoid cell lines from BWCFF patients with different ACTB or ACTG1 mutations demonstrated distinct abnormal patterns of F-actin organization, suggesting that the morphological impact of missense mutations may differ in a mutation-specific way.7 The in vitro and in vivo effects of single mutations have been studied for eight ACTG1 mutations causing DFNA20/26:32, 33 each mutation leads to specific alteration of the cell morphology, actin polymerization and interaction with the actin-binding proteins. An experiment with two BWCFF-associated ACTB mutations (p.Arg196His and p.Asn12Asp) confirmed mutation-specific morphological changes.7 Procaccio et al. showed that p.Arg183Trp affects the ATP-binding pocket resulting in a more stable actin polymer, which is not depolymerized by the severing protein cofilin. This particular mutation may have a functional effect on development similar to other BWCFF variants, but may interfere specifically with another cellular process to explain the dystonia. Johnson et al9 showed that the ACTB p.Glu117Lys variant alters cell adhesion to a fibronectin-coated surface, reduces formation of protrusive structures and modify polymer formation properties.
A p.Glu364Lys mutation has been reported in a patient with intellectual disability, photosensitivity and recurrent infections resulting from the impaired neutrophil chemotactic response.34 Dysmorphology was not addressed. This mutation did not affect in vitro actin polymerization. It occurred in a region of the protein important for profilin binding, suggesting that this alteration may have very different physiopathologic consequences. From Nunoi’s report, it is not possible to determine whether this patient had a phenotype compatible with BWCFF.
Nosology
Patients with BWCFF exhibit marked phenotypic variability, which probably explains why they were reported as distinct clinical entities before the discovery of actin involvement. Several non-genotyped patients in the literature could have BWCFF,35, 36, 37, 38 one of them carrying an inv(2)(p12q14)mat.39 FA was originally thought to be a distinct entity,4 but their patient 1 has the recurrent ACTB p.Arg196His mutation (B10), and another FA patient with duplicated halluces40 had the recurrent ACTB mutation p.Arg196Cys (B24). FA appears to represent the aged phenotype of BWCFF with coarsened traits (Figure 4). Fryns–Aftimos’ and Der Kaloustian’s cases5 were classified by Winter as CFF3. We have shown here that some CFF3 patients41, 42 carry an ACTB mutation.
Cerebro-oculo-facial-lymphatic syndrome,43 and some patients with normal intelligence reported as Teebi hypertelorism syndrome may have BWCFF.44 Pashayan et al45, 46 reported a dominant blepharo-naso-facial syndrome: dysmorphology is different but torsion dystonia and deafness represent intriguing similarities with actinopathies.47 Mutation-negative patient 5 illustrating Kabuki syndrome gene discovery48 strikingly resembles BWCFF.
Similarities between Noonan syndrome and BWCFF were discussed by Verloes.49 This diagnosis has been suggested in infancy for several patients from our series. In mild cases, the facial appearance in infancy (when metopic ridge is absent), chest deformity and nuchal skinfolds or pterygium colli are confounding (cf. B11 or B16).
Conclusion
The story of BWCFF nicely illustrates how new molecular diagnoses are redefining and expanding the clinical spectrum of what we previously consider as distinct syndromes, and how NGS technologies, by unraveling the molecular basis of clinically defined entities, will reorganize nosology for many developmental disorders.
BWCFF is a complex developmental disorder with craniofacial, visceral and muscular involvement due to gain-of-function mutations in ACTB and ACTG1, illustrating the wide-ranging effect of dysregulating cytoplasmic actin function in cranial neural crest derivatives during development. Although minimal criteria for clinical diagnosis are still difficult to settle, the facial phenotype seems the most reliable handle at all ages. Neural migration defect and ocular colobomata are highly suggestive, but not mandatory. In young patients with mild BWCFF, the Gestalt overlaps with Noonan syndrome (e.a. ptosis, CHD, and pectus) but pulmonary stenosis was not seen in BWCFF. Arched eyebrows and wide but short nose are probably the best handle to differentiate the two syndromes. Coloboma is rarely seen in Noonan syndrome, and neuronal migration defects are not a feature: presence of any of these signs should prompt for ACT screening. Euryblepharon may also falsely point to Kabuki syndrome.
The presence of hematologic malignancies in two of our cases (one with a mutation in ACTG1 and one in ACTB) may not be incidental. Although our data set is not large enough to state that BWCFF is associated with an increased tumor risk, there may be predisposition, particularly to hematological malignancies.
We have collected a few convincing BWCFF cases without a variation of the coding sequence of either ACTB or ACGT1. Considering the number of actin-binding protein partners of these two ubiquitous actins, further genetic heterogeneity of BWCFF is a reasonable hypothesis.
Accession codes
References
Baraitser M, Winter RM : Iris coloboma, ptosis, hypertelorism, and mental retardation: a new syndrome. J Med Genet 1988; 25: 41–43.
Ramer JC, Mascari MJ, Manders E, Ladda RL : Syndrome identification # 149: trigonocephaly, pachygyria, retinal coloboma and cardiac defect: a distinct syndrome. Dysmorph Clin Genet 1992; 6: 15–20.
Ramer JC, Lin AE, Dobyns WD et al: Previously apparently undescribed syndrome: shallow orbits, ptosis, coloboma, trigonocephaly, gyral malformations, and mental and growth retardation. Am J Med Genet 1995; 57: 403–409.
Fryns JP, Aftimos S : New MCA/MR syndrome with distinct facial appearance and general habitus, broad and webbed neck, hypoplastic inverted nipples, epilepsy and pachygyria of the frontal lobes. J Med Genet 2000; 37: 460–462.
Winter RM : Cerebro-fronto-facial syndrome: three types? Clin Dysmorphol 2001; 10: 79–80.
Koçak Eker H, Ünal S, Derinkuyu BE, Masliah-Planchon J, Drunat S, Verloes A : Cerebro-fronto-facial syndrome type 3: a clinical presentation of Baraitser-Winter syndrome. Eur J Med Genet 2013; 57: 32–36.
Riviere JB, van Bon BW, Hoischen A et al: De novo mutations in the actin genes ACTB and ACTG1 cause Baraitser-Winter syndrome. Nat Genet 2012; 44: S441–S442.
Di Donato N, Rump A, Koenig R et al: Severe forms of Baraitser-Winter syndrome are caused by ACTB mutations rather than ACTG1 mutations. Eur J Hum Genet 2013; 22: 179–183.
Johnston JJ, Wen KK, Keppler-Noreuil K et al: Functional analysis of a de novo ACTB mutation in a patient with atypical Baraitser-Winter syndrome. Hum Mutat 2013; 34: 1242–1249.
Gearing M, Juncos JL, Procaccio V et al: Aggregation of actin and cofilin in identical twins with juvenile-onset dystonia. Ann Neurol 2002; 52: 465–476.
Procaccio V, Salazar G, Ono S et al: A mutation of beta -actin that alters depolymerization dynamics is associated with autosomal dominant developmental malformations, deafness, and dystonia. Am J Hum Genet 2006; 78: 947–960.
Pollard TD, Cooper JA : Actin, a central player in cell shape and movement. Science 2009; 326: 1208–1212.
Schoenenberger CA, Mannherz HG, Jockusch BM : Actin: from structural plasticity to functional diversity. Eur J Cell Biol 2011; 90: 797–804.
Bergeron SE, Zhu M, Thiem SM, Friderici KH, Rubenstein PA : Ion-dependent polymerization differences between mammalian beta- and gamma-nonmuscle actin isoforms. J Biol Chem 2010; 285: 16087–16095.
Dugina V, Zwaenepoel I, Gabbiani G, Clement S, Chaponnier C : Beta and gamma-cytoplasmic actins display distinct distribution and functional diversity. J Cell Sci 2009; 122: 2980–2988.
Khaitlina SY : Functional specificity of actin isoforms. Int Rev Cytol 2001; 202: 35–98.
Kilpinen S, Autio R, Ojala K et al: Systematic bioinformatic analysis of expression levels of 17,330 human genes across 9,783 samples from 175 types of healthy and pathological tissues. Genome Biol 2008; 9: R139.
Hill MA, Gunning P : Beta and gamma actin mRNAs are differentially located within myoblasts. J Cell Biol 1993; 122: 825–832.
Bassell GJ, Zhang H, Byrd AL et al: Sorting of beta-actin mRNA and protein to neurites and growth cones in culture. J Neurosci 1998; 18: 251–265.
Karakozova M, Kozak M, Wong CC et al: Arginylation of beta-actin regulates actin cytoskeleton and cell motility. Science 2006; 313: 192–196.
Bunnell TM, Burbach BJ, Shimizu Y, Ervasti JM : beta-Actin specifically controls cell growth, migration, and the G-actin pool. Mol Biol Cell 2011; 22: 4047–4058.
Bulinski JC : Cell biology. Actin discrimination. Science 2006; 313: 180–181.
Tondeleir D, Vandamme D, Vandekerckhove J, Ampe C, Lambrechts A : Actin isoform expression patterns during mammalian development and in pathology: insights from mouse models. Cell Motil Cytoskeleton 2009; 66: 798–815.
Lloyd CM, Berendse M, Lloyd DG, Schevzov G, Grounds MD : A novel role for non-muscle gamma-actin in skeletal muscle sarcomere assembly. Exp Cell Res 2004; 297: 82–96.
Rybakova IN, Patel JR, Ervasti JM : The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J Cell Biol 2000; 150: 1209–1214.
Sonnemann KJ, Fitzsimons DP, Patel JR et al: Cytoplasmic gamma-actin is not required for skeletal muscle development but its absence leads to a progressive myopathy. Dev Cell 2006; 11: 387–397.
Guo C, Liu S, Wang J, Sun MZ, Greenaway FT : ACTB in cancer. Clin Chim Acta 2013; 417: 39–44.
Lohr JG, Stojanov P, Lawrence MS et al: Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci USA 2012; 109: 3879–3884.
Batista FD, Treanor B, Harwood NE : Visualizing a role for the actin cytoskeleton in the regulation of B-cell activation. Immunol Rev 2010; 237: 191–204.
Oike T, Ogiwara H, Nakano T, Yokota J, Kohno T : Inactivating mutations in SWI/SNF chromatin remodeling genes in human cancer. Jpn J Clin Oncol 2013; 43: 849–855.
Miyamoto K, Gurdon JB : Transcriptional regulation and nuclear reprogramming: roles of nuclear actin and actin-binding proteins. Cell Mol Life Sci 2013; 70: 3289–3302.
Morin M, Bryan KE, Mayo-Merino F et al: In vivo and in vitro effects of two novel gamma-actin (ACTG1) mutations that cause DFNA20/26 hearing impairment. Hum Mol Genet 2009; 18: 3075–3089.
Bryan KE, Rubenstein PA : Allele-specific effects of human deafness gamma-actin mutations (DFNA20/26) on the actin/cofilin interaction. J Biol Chem 2009; 284: 18260–18269.
Nunoi H, Yamazaki T, Tsuchiya H et al: A heterozygous mutation of beta-actin associated with neutrophil dysfunction and recurrent infection. Proc Natl Acad Sci USA 1999; 96: 8693–8698.
Schaap C, Schrander-Stumpel C, Fryns JP : Opitz C syndrome: on the nosology of mental retardation and trigonocephaly. Genet Couns 1992; 3: 209–215.
Fryns JP : Previously apparently undescribed syndrome: shallow orbits, ^ptosis, coloboma, trigonocephaly, gyral malformations, and mental and growth retardation. Am J Med Genet 1996; 64: 521–522.
Ganesh A, Al-Kindi A, Jain R, Raeburn S : The phenotypic spectrum of baraitser-winter syndrome: a new case and review of literature. J AAPOS 2005; 9: 604–606.
Shiihara T, Maruyama K, Yamada Y et al: A case of Baraitser-Winter syndrome with unusual brain MRI findings: pachygyria, subcortical-band heterotopia, and periventricular heterotopia. Brain Dev 2010; 32: 502–505.
Pallotta R : Iris coloboma, ptosis, hypertelorism, and mental retardation: a new syndrome, possibly localised on chromosome 2. J Med Genet 1991; 28: 342–344.
Der Kaloustian VM, Pelletier M, Costa T, Blackston DR, Oudjhane K : A new syndrome with craniofacial and skeletal dysmorphisms and developmental delay. Clin Dysmorphol 2001; 10: 87–93.
Guion-Almeida ML, Richieri-Costa A : Acrocallosal syndrome: report of a Brazilian girl. Am J Med Genet 1992; 43: 938–941.
Guion-Almeida ML, Richieri-Costa A : Frontonasal dysplasia, macroblepharon, eyelid colobomas, ear anomalies, macrostomia, mental retardation and CNS structural anomalies: defining the phenotype. Clin Dysmorphol 2001; 10: 81–86.
Milunsky JM, Capin DM : Cerebro-oculo-facial-lymphatic syndrome. Clin Genet 2003; 63: 1–6.
Machado-Paula LA, Guion-Almeida ML : Teebi hypertelorism syndrome: additional cases. Am J Med Genet 2003; 117A: 181–183.
Putterman AM, Pashayan H, Pruzansky S : Eye findings in the blepharo-naso-facial malformation syndrome. Am J Ophthalmol 1973; 76: 825–831.
Pashayan H, Pruzansky S, Putterman A : A family with blepharo-naso-facial malformations. Am J Dis Child 1973; 125: 389–393.
Sachdev M, Rastogi A, Singh A et al: Phenotypic overlap between Blepharo-naso-facial syndrome and Nablus mask-like syndrome. Report from the first Indian family. Ophthalmic Genet 2013; 34: 65–68.
Ng SB, Bigham AW, Buckingham KJ et al: Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet 2010; 42: 790–793.
Verloes A : Iris coloboma, ptosis, hypertelorism, and mental retardation: Baraitser-Winter syndrome or Noonan syndrome? J Med Genet 1993; 30: 425–426.
Bitton N, Alexander S, Ruggiero S, Parameswaran A, Russo A, Ferguson F : Case report: Noonan-like multiple central giant cell granuloma syndrome. Pediatr Dent 2012; 34: 144–147.
Rossi M, Guerrini R, Dobyns WB, Andria G, Winter RM : Characterization of brain malformations in the Baraitser-Winter syndrome and review of the literature. Neuropediatrics 2003; 34: 287–292.
Acknowledgements
We would like to thank all the patients and their families for their interest and participation. This work was supported by the Baily Thomas Charitable Fund, UK and the National Institute for Social Care and Health Research (NISCHR), Wales, UK.
DEDICATION
This paper is dedicated to Professor Michael Baraitser and to the memory of Professor Robin Winter, who first described patients with a condition, which was subsequently named ‘Baraitser–Winter syndrome’. Dysmorphology has been vastly enriched by their contributions to this discipline.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Verloes, A., Di Donato, N., Masliah-Planchon, J. et al. Baraitser–Winter cerebrofrontofacial syndrome: delineation of the spectrum in 42 cases. Eur J Hum Genet 23, 292–301 (2015). https://doi.org/10.1038/ejhg.2014.95
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.95
This article is cited by
-
Continuous high-frequency deep brain stimulation of the anterior insula modulates autism-like behavior in a valproic acid-induced rat model
Journal of Translational Medicine (2022)
-
Obsessive–compulsive symptoms in ACTG1-associated Baraitser-Winter cerebrofrontofacial syndrome
Journal of Neural Transmission (2022)
-
Dystonia-deafness syndrome caused by ACTB p.Arg183Trp heterozygosity shows striatal dopaminergic dysfunction and response to pallidal stimulation
Journal of Neurodevelopmental Disorders (2018)
-
Poland’s Syndrome: Are Postzygotic Mutations in β-Actin Associated with its Pathogenesis?
American Journal of Clinical Dermatology (2018)
-
Variants in exons 5 and 6 of ACTB cause syndromic thrombocytopenia
Nature Communications (2018)
