
Abstract
Isolated cytochrome c oxidase (COX) deficiency is a prevalent cause of mitochondrial disease and is mostly caused by nuclear-encoded mutations in assembly factors while rarely by mutations in structural subunits. We hereby report a case of isolated COX deficiency manifesting with encephalomyopathy, hydrocephalus and hypertropic cardiomyopathy due to a missense p.R20C mutation in the COX6B1 gene, which encodes an integral, nuclear-encoded COX subunit. This novel mutation was predicted to be severe in silico. In accord, enzymatic activity was undetectable in muscle and fibroblasts, was severely decreased in lymphocytes and the COX6B1 protein was barely detectable in patient’s muscle mitochondria. Complementation with the wild-type cDNA by a lentiviral construct restored COX activity, and mitochondrial function was improved by 5-aminoimidazole-4-carboxamide ribonucleotide, resveratrol and ascorbate in the patient’s fibroblasts. We suggest that genetic analysis of COX6B1should be included in the investigation of isolated COX deficiency, including patients with cardiac defects. Initial measurement of COX activity in lymphocytes may be useful as it might circumvent the need for invasive muscle biopsy. The evaluation of ascorbate supplementation to patients with mutated COX6B1 is warranted.
Similar content being viewed by others


INTRODUCTION
Inherited mitochondrial diseases affecting OXPHOS function are prevalent with an estimated incidence of 1:5000. They are heterogeneous; symptoms may present at any age and may affect a wide range of tissues, although those with high energy demands, such as the brain, muscle and liver are preferentially affected. The underlying molecular defect may arise either from mutations in the nuclear or the mitochondrial genome and may cause isolated or combined defects in the mitochondrial respiratory chain complexes. Generally, most nuclear-encoded defects present earlier as devastating multisystemic disorders.1 Among these, isolated cytochrome c oxidase (COX), mitochondrial respiratory chain complex IV, EC 1.9.3.1 deficiency (MIM 220110) is a prevalent cause and is mostly associated with severe encephalomyopathy and Leigh syndrome (LS).2
COX, localized in the inner mitochondrial membrane, is the final electron acceptor in the mitochondrial electron transport chain. This multisubunit copper-heme oxidase transfers electron from reduced cytochrome c to molecular oxygen while translocating protons from the mitochondrial matrix to the intermembrane space. COX functions as a dimer, and the monomer is composed of 13 subunits. The three catalytic, larger subunits are encoded in the mitochondrial genome (mtDNA) while the remaining are nuclear encoded. The biogenesis and assembly of COX depends on numerous ancillary factors, including copper chaperones, all nuclear encoded. The majority of isolated COX deficiencies are caused by mutations in these factors.2 Specifically, disease-causing mutations were found in genes encoding SURF1, essential for the formation of early assembly intermediates; SCO1 and SCO2, required for the insertion of the copper moiety; COX10 and COX15, essential for heme A biosynthesis; and finally in LRPPRC and TACO1, essential for COX expression.2, 3
To date, only three structural subunits have been associated with disease; we reported a mutation in COX4I2 in a patient with exocrine pancreatic insufficiency,4 Indrieri et al reported an unconventional mitochondrial disease characterized by microphthalmia with linear skin lesions caused by a COX7B defect and Massa et al described a mutation in the nuclear-encoded COX6B1 in a case of infantile encephalomyopathy.5, 6 We hereby present the second reported case of mutated COX6B1 due to a novel mutation, while expanding the clinical spectrum to include cardiomyopathy, and evaluating various therapeutic small molecules.
SUBJECT AND METHODS
Subject
The affected individual, a male, was the fourth child born to an Arab Muslim family of Palestinian origin. Parents are first cousins. One sibling, a girl died at the age of 1 month due to SIDS according to parents, without any investigation.
Informed consent for muscle, skin biopsy and exome sequencing was obtained from the parents, and the study was approved by the local institutional review board.
During pregnancy, the patient was suspected to have IUGR and was delivered by Cesarian section at 36+2 gestational weeks with Apgar scores 9 and 10. Birth weight was 2.3 kg (10th percentile) and head circumference 32 cm (10th percentile). Physical exam after birth, detected a systolic murmur 3/6.
On the first day of life, the patient developed metabolic acidosis with lactate level of 27 mM (normal range 0.5–2.4 mM) and elevated ammonia levels 277 μ M (normal range 10–60 μ M). Metabolic work up showed normal acylcarnitines, elevated alanine level 1400 μ M (normal <700 μ M), and urine organic acids showed increased lactic acidosis and ketones.
Chest X-ray revealed cardiomegaly, and cardiac Echo revealed symmetrical left ventricular hypertrophy tricuspid regurge and pulmonary hypertension. The patient was transported to the NICU and treated with bicarbonate, acetate and diuretics. At the age of 4 days, quadriceps muscle biopsy was performed. Pathology showed decreased cytochrome c oxidase stain.
Brain ultrasound performed at age 5 days was normal. Subsequently, head circumference grew rapidly adding 13 centimeters by age 35 days, repeated brain US revealed dilated ventricles confirmed by head CT, which also showed subcortical and white matter cortical hemorrhage in the occipital region. At 15 weeks, a ventriculoperitoneal (VP) shunt was inserted. Head CT performed at age 2 years showed prominent dilatation of the ventricles (Figure 1). Due to hypotonia with feeding difficulties and recurrent aspirations, a percutaneous endoscopic gastrostomy (PEG) feeding tube was inserted at age 10 weeks. Eye examination at the age of 15 weeks revealed cortical blindness. Hearing test ABR was normal.

Head CT. Head CT performed at age 2 years shows prominent dilatation of the ventricles, VP shunt in the right occipital region, and of note is the very thin cortex.
Repeated cardiac Echo continued to show mild tricuspid regurge, mild left ventricular hypertrophy (LVH) and pulmonary hypertension (PHTN) in the first months of life. At the age of 4 months, he started developing hypertrophic obstructive cardiomyopathy, he was started on diuretics and beta blockers and later calcium channel blockers were added; follow-up Echo at the age of 2 years revealed improvement in the LVH and no PHTN.
Coenzyme Q10 supplementation (60 mg twice daily) was initiated at 4 months but discontinued by the parents. In the following months, the patient was admitted several times to the hospital mainly due to chest infections, shunt malfunctioning and fevers. He died at the age of 30 months due to severe hypoxemia after a febrile illness with chest infection and pulmonary edema; parents refused mechanical ventilation and refused reanimation.
Enzymatic assays
Enzymatic activities of rotenone-sensitive NADH CoQ reductase (Complex I), succinate cytochrome c reductase (complex II+III), succinate dehydrogenase (complex II, SDH), COX (complex IV) and citrate synthase (CS), a mitochondrial marker enzyme, were determined in isolated muscle mitochondria, lymphocyte and fibroblast homogenates as we have previously described.7, 8 All spectrophotometric measurements were carried out using a double-beam spectrophotometer (UVIKON 930, Secomam, France).
Western blotting analysis
Western blotting analysis was performed after separation of mitochondrial proteins by Urea–sodium dodecylsulfate-polyacrylamide gel electrophoresis (Urea-SDS-PAGE) essentially as described9 with the following modifications; subsequently to electrophoresis (12% separating and 5% stacking gels), proteins were transferred to a polyvinylidene fluoride membrane, blocked with protein-free blocking buffer (Thermo scientific, Rockford, IL, USA) and sequentially exposed to primary antibodies: anti COX6B1 (Sigma-Aldrich, St Louis, MO, USA), anti-MTCO1, anti-Complex IV subunit II, anti-complex II 70 KDa subunit (succinate dehydrogenase, SDH), (MitoSciences, Eugene, OR, USA) overnight. Subsequently, the membrane was incubated with peroxidase-conjugated secondary antibodies (Jackson Immuno Research laboratories,West Grove, PA, USA) and visualized by chemiluminescence detection using the EZ-ECL kit (Biological Industries, Kibbutz Beit Ha’emek, Israel). Calculation of band intensities was made by the ImageJ program, http://imagej.nih.gov/ij (National Institute of Health, Bethesda, MD, USA).
Structural analysis of the effect of R20C mutations
The solved structure of bovine cytochrome c oxidase10 was inspected to evaluate the structural role of R20 in COX6B1, as well as to assess the effects on COX6B1 stability and activity of the mutation found in this study, p.R20C in COX6B1, and of the previously reported mutation at the same position, p.R20C (p.R19H when numbering without the first M).6 These mutations were introduced in silico onto the COX6B1 subunit (chain H of 2eij), and their effect on stability was modeled using the Rosetta (release 3.5) DDG_monomer application.11 The running parameters of protocol 1 was used (ie only the mutated chain was allowed to move), with the following command line for, for example, the R19C mutation: ddg_monomer.linuxgccrelease -database rosetta_database -s 2eijH.pdb -resfile resfileR19C -ddg::weight_file soft_rep_design -ddg::iterations 1 -ddg::local_opt_only true -in::file::fullatom -ddg::min_cst false -ddg::mean false -ddg::min true -ddg::sc_min_only false -ddg::opt_radius 0.1 -scorefile score_R19C.sc).
Figure 2 was produced using pymol (http://pymol.org/).

Structure-based assessment of effect the R20C and R20H mutations in the COX6B1 protein. (a) Structure of the wild-type protein (PDB id 2eij).6, 10, 11 Note the major contribution of R20 to a network of hydrogen bonds involving residues D18-R20-D36-R39 that orients the n-terminal tail (including residues D18 and D20) relative to the adjacent helix (including D36 and R39). The local area shown in the following panels is highlighted by a square. (b) In order to assess the effects of the mutations R20C (from this study) and R20H,6 respectively, these mutations were introduced in silico: Wild type and mutations are shown in the same one figure (upper panel), and for better clarity, also in separate panels for R20C (middle panel) and R20H (lower panel). Notably, neither histidine nor cysteine are able to form adequate hydrogen bonds, owing to both size and orientation. However, the effect of cysteine is predicted to be more dramatic due to loss of positive charge.
Whole-exome sequencing
Whole-exome sequencing was performed on DNA from the patient using the SureSelect Human All Exon V.2 Kit (Agilent Technologies, Santa Clara, CA, USA) on HiSeq2000 (Illumina, San Diego, CA, USA) as 100-bp paired-end runs. Reads alignment and variant calling were performed with the DNAnexus software (DNAnexus, Palo Alto, CA, USA; https://www.dnanexus.com/) using the default parameters, with the human genome assembly hg19 (GRCh37) as a reference. The data obtained were submitted to the Mitochondrial Disease Sequence Data Resource (MSeqDR) Consortium https://mseqdr.org/index.php.
Tissue culture and evaluation of compounds
Patient and control fibroblasts obtained from forearm skin biopsies were cultured in DMEM medium containing 4.5 g/l glucose supplemented with 15% fetal calf serum (FCS), 1% penicillin–streptomycin, 365 μg/ml l-glutamine, 110 μg/ml pyruvate and 50 μg/ml uridine (GLU, permissive medium) or in glucose-free (GAL) DMEM supplemented with 10% dialyzed FCS, 1% penicillin–streptomycin, 365 μg/ml l-glutamine and 5 mM galactose (GAL, restrictive medium) at 37 °C in 5% CO (all cell culture media and solutions were purchased from Biological Industries).
Evaluation of compound was carried out essentially as we have previously described in 96-well microtiter plates.12 Briefly, growth was evaluated by methylene blue and ATP content was determined by the ATPlite luminescence assay system according to the manufacturer’s instructions (Perkin Elmer, Waltham, MA, USA) after 72 h incubation in GLU or GAL medium, respectively. Mitochondrial content was estimated by MitoTracker Green FM (MTG) and membrane potential by tetramethylrhodamine ethyl ester (TMRE) (Molecular Probes, Eugene, OR, USA). Briefly, 200 nM MitoTracker Green was added to the medium for 45 min and 50 nM TMRE was added successively for an additional 45 min. The medium was removed, washed and replaced with phosphate-buffered saline before fluorescence readings at λex 485 nm, λem 528 nm (MTG) and λex 485 nm, λem 590 nm (TMRE). The following compounds were evaluated: 0.5 mM 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) (Tocris Bioscience, Bristol, UK), 10 μ M ascorbate; 0.1 mM bezafibrate; 4 mM N-acetylcysteine (NAC) 25 μ M resveratrol (Sigma-Aldrich, Rehovot, Israel); or vehicle dimethyl sulfoxide (DMSO).
Data were analyzed by the two-tailed Student’s t-test and one-way ANOVA using the SPSS version 20 program (IBM corp., Armonk, NY, USA). P<0.05 was regarded as statistically significant.
Wild-type (wt) cDNA complementation.
wt C6ORF66 cDNA was cloned into the pLenti6/V5-D-TOPO 6969-bp expression vector by directional TOPO cloning (primers available upon request). The recombinant vector was propagated in One Shot Stbl3 E. coli on ampicillin and introduced into 293FT cells after co-transfection with pLP1, pLP2 and pLP/VSVG plasmids with Lipofectamine-2000, according to the manufacturer’s instructions (Vira-Power, Lentiviral expression system, Invitrogen, Life Technologies, Grand Island, NY, USA). Patient’s fibroblasts were infected overnight with the lentiviral construct in the presence of polybrene (Sigma-Aldrich, Rehovot, Israel), and stably transduced cells were established in 10 days by blasticin selection (Invitrogen, Life Technologies).
RESULTS
Biochemical studies
The enzymatic analysis of mitochondrial respiratory chain complexes in muscle mitochondria revealed undetectable COX (complex IV) activity, whereas complexes I, II and II+III were within the normal limit. A complex IV defect was also evident in lymphocyte homogenates but to a lesser extent, with 17% residual activity normalized to CS, while complex II was within the normal range (Table 1).
Based on these findings and in accord with histochemistry, the diagnosis of isolated COX deficiency was made, and molecular investigation was initiated.
Molecular analysis
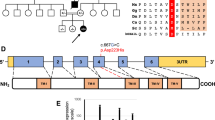
Exome analysis yielded 50,373,369 reads which were confidently aligned to the human reference assembly HG19. In all, 136,447 variants were called and filtered, removing those which were heterozygous, on the X-chromosome, synonymous, low-covered (< × 7), off target and those present in dbSNP132 or the in-house dbSNP. Twelve variants survived but only one of them resided in a gene encoding a mitochondrial protein; in COX6B1, a missense mutation was detected in hg19_chr19: g.36142203C>T NM_001863.4:r.58c>u (ENST00000246554 c.58C>T) changing the Arg at position 20 to Cys in the protein (NP_001854.1:p.R20C).
The residue is conserved in mammals, Drosophila melongaster and Caenorhabditis elegans. This transition mutation was rated as disease causing by Mutation Taster (amino-acid change score 4.91) (http://www.mutationtaster.org/) and probably damaging by PolyPhen-2 (score 0.89) (http://genetics.bwh.harvard.edu/pph2/).
The mutation was not present in the 6503 exome analyses of healthy individuals available through the Exome Variant Server, NHLBI Exome Sequencing Project, Seattle, Washington, USA (http://evs.gs.washington.edu) (EVS-v.0.0.21; 18 October 2013), and Sanger sequencing of parents and two healthy siblings confirmed segregation with the disease.
Analysis in silico of potential impact on structural stability of COX6B1 R20C and R20H mutations
Arginine R20 is highly conserved throughout evolution and is therefore expected to play an important role in the function and/or structure of COX6B1. Arginine residue R20 is a central part of an extended network of hydrogen bonds involving D18-R20-D36-R39, which orients the flexible n-terminal tail by connecting it to an adjacent helix (Figures 2a and b, upper panel). The mutation of R20 to cysteine in the present study (Figure 2b, middle panel) has a predicted stronger effect (R20C: ΔΔGfold predicted=0.86 kcal/mol) than a corresponding previously reported mutation to the characteristically more similar Histidine (R20H: ΔΔGfold predicted=0.45 kcal/mol; Figure 2b, lower panel).6, 10, 11
Although both of these predictions anticipate only minor effects on protein stability (only values ΔΔGfold predicted>1.00 are usually defined as stability hotspots), it can be anticipated that both mutations will have stronger effects owing to more delocalized effects, resulting from disruption of this extended network of hydrogen bonds that might not be fully captured by our calculations (Figure 2b).
Western blotting analysis
Urea-PAGE combined with western blotting analysis of patient’s muscle mitochondrial revealed a barely detectable residual amount of COX6B1 protein (11% of control normalized to SDH), suggesting that the mutant protein in highly unstable.
In addition, COX subunits I and II were also decreased but to a much lesser extent (60% and 40% of control, respectively; Figure 3).

Western blotting analysis. Equal amount of protein (8 μg) containing equal amount of CS activity (3.5 mU), from control (C) and patient (P) muscle mitochondria was separated by Urea-SDS-PAGE and subjected to western blotting with primary antibodies: anti COX6B1, anti-MTCO1 (COX I), anti-Complex IV subunit II (COX II), anti-complex II 70 KDa subunit (SDH). These were subsequently detected by peroxidase conjugated secondary antibodies and visualized by chemiluminescence (a). Band intensities quantified depicted in the graph relatively to SDH showed severely decreased COX6B1 and partially decreased COX II and COX I (b).
Complementation with wt COX6B1
Patient’s fibroblasts grown on GLU were stably transfected with a lentiviral construct containing the wtCOX6B1 cDNA gene or a control vector. COX activity was undetectable in the control-transfected cells, whereas COX6B1 restored activity to levels near normal fibroblasts range, confirming the pathogenicity of the p.R20C mutation. Notably, CS activity decreased with increased COX activity, whereas complex II remained constant (Table 2). The restoration was also reflected in the normalized ATP content and the ability to maintain grow on GAL medium (Figure 4).

The effect of small molecules on patient’s fibroblasts. Control (C) fibroblasts (n=5), patient (P) fibroblasts or patient fibroblast transfected with wt gene (P+COX6B1) were seeded and grown in GLU or GAL medium. Patient’s fibroblasts were also grown on GAL in the presence of either of the following compounds: AICAR; ascorbate; bezafibrate; N-acetylcysteine (NAC) or resveratrol or vehicle (no additive). Cell growth (a) was measured by methylene blue at 620 nm (A620). ATP content (b) was measured by luciferin-luciferase and normalized to growth. Values are presented as normalized mean±SEM. $ Statistically significant P<0.05, when compared with normal control. Asterisk (*) statistically significant when compared with untreated patient cells.
Evaluation of small molecules
The mitochondrial dysfunction was evident in the patient’s fibroblasts where defective growth and ATP content were clearly observed in GAL medium as compared with glucose-containing (GLU) medium. Thus the effect of mitochondrial biogenensis inducers (AICAR, bezafibrate, resveratrol) and antioxidants (ascorbate, NAC) was evaluated on GAL medium. The concentrations were chosen according to literature and our previous experience (Figure 4).13
All tested compounds slightly increased growth relative to untreated cells. ATP content was markedly improved by AICAR, reveratrol and ascorbate. The favorable response is presumably not due to increased mitochondrial content or improved membrane potential as mitotracker green and TMRE stain did not reveal any significant differences between treated and untreated cells (result not shown).
DISCUSSION
This the first report of an isolated COX deficiency due a novel p.R20C mutation in COX6B1 and the second report of a mutation in this gene and residue. This mutation was predicted to be disease causing, and the COX activity in the patient’s fibroblasts was restored by complementation with the wt cDNA confirming its pathogenicity. Interestingly, the same Arg residue mutated in our patient is the same one as was previously reported by Massa et al6 who also demonstrated defective COX assembly. However, the amino-acid changes are different; in the present case, the change is to Cys instead of to His, indicating that most probably the R20 residue is a hotspot. Inspection of the COX6B1 structure reveals that this residue is important for the stabilization of the protein, as already suggested.6, 10 R20 is involved in an extended network of hydrogen bonds, and the predicted reduction of stability due to the disruption of these bonds is higher for the Cys change than for the His. The R20C mutation seems, indeed, to have a more severe effect also in vivo as COX activity in muscle and fibroblasts was undetectable and the COX6B1 protein was severely reduced in muscle, whereas in the previously reported case some residual activity was observed in these tissues and the COX6B1 was apparently present. Notably, the biochemical defect was also present in lymphocytes, albeit to a lesser degree, than in the muscle (Table 1).
In accord with the biochemical findings, our patient also declined more rapidly and succumbed earlier (at 2.5 years compared with 10 years6). The clinical features were also different, including a combination of encephalomyopathy with hydrocephalus and hypertropic cardiomyopathy, thereby expanding the clinical spectrum of COX6B1 defects. Selective involvement of non-neural tissues combined with encephalopathy and Leigh diseases has previously been observed in isolated COX deficiencies, particularly in conjunction with COX assembly gene mutations, such as SCO2.2, 14 Accordingly, COX1B should be included in the panel of genes examined in isolated COX encephalomyopathies with or without cardiac involvement.
With the exception of reversible COX deficiencies due to TRM15 and mt-tRNAglu,16 isolated COX deficiency is a devastating disease. Currently, no satisfactory treatment is available. The EPI-743 treatment was beneficial to patients with LS due to mutations in SURF1 and in LS with secondary COX deficiency due to mutated ETHE1.17 In vitro studies in patient fibroblasts showed that copper18 and copper with bezafibrate supplementation rescued cells harboring mutations in SCO2 encoding a COX copper-chaperone19 while L-cystein supplementation improved mitochondrial function in cells with TRMU mutations.20 However, as the COX6B1 defect does not cause LS and the protein is not a copper chaperone but an integral COX subunit, we evaluated a number of other potentially beneficial molecules for their ability to improve mitochondrial function in the patient’s fibroblasts. The significantly beneficial effect of AICAR on ATP content, and resveratrol on both ATP content and growth resembles the effect of these mitochondrial biogenesis inducers, on defects due to GFM1 and MRPS22, mutations in the mitochondrial translation machinery.12 Notably, also ascorbate had a significant positive effect on both parameters without increasing mitochondrial content similar to the effect on cells with mutated mitochondrial elongation factor Ts. This effect could be due to its antioxidant properties21 or its ability to donate electrons via cytochrome c. As vitamin C is regarded as a safe food supplement, it could be administered to patients. Regretfully, we were not able to apply this knowledge in our patient as he succumbed before the results of this study were obtained.
To summarize, we suggest that sequencing of COX6B1 should be included in the investigation of isolated COX deficiency, including patients with cardiac defects. As this defect is expressed in lymphocytes, the initial measurement of COX activity in blood could in some cases, and possibly also in other mitochondrial diseases, circumvent the need for a more invasive muscle biopsy.22 In the future, the evaluation of vitamin C supplementation in patients is warranted.
References
Di Donato S : Multisystem manifestations of mitochondrial disorders. J Neurol 2009; 256: 693–710.
DiMauro S, Tanji K, Schon EA : The many clinical faces of cytochrome c oxidase deficiency. Adv Exp Med Biol 2012; 748: 341–357.
Soto IC, Fontanesi F, Liu J, Barrientos A : Biogenesis and assembly of eukaryotic cytochrome c oxidase catalytic core. Biochim Biophys Acta 2012; 1817: 883–897.
Shteyer E, Saada A, Shaag A et al: Exocrine pancreatic insufficiency, dyserythropoeitic anemia, and calvarial hyperostosis are caused by a mutation in the COX4I2 gene. Am J Hum Genet 2009; 84: 412–417.
Indrieri A, van Rahden VA, Tiranti V et al: Mutations in COX7B cause microphthalmia with linear skin lesions, an unconventional mitochondrial disease. Am J Hum Genet 2012; 91: 942–949.
Massa V, Fernandez-Vizarra E, Alshahwan S et al: Severe infantile encephalomyopathy caused by a mutation in COX6B1, a nucleus-encoded subunit of cytochrome c oxidase. Am J Hum Genet 2008; 82: 1281–1289.
Saada A, Bar-Meir M, Belaiche C, Miller C, Elpeleg O : Evaluation of enzymatic assays and compounds affecting ATP production in mitochondrial respiratory chain complex I deficiency. Anal Biochem 2004; 335: 66–72.
Reisch AS, Elpeleg O : Biochemical assays for mitochondrial activity: assays of TCA cycle enzymes and PDHc. Methods Cell Biol 2007; 80: 199–222.
Darley-Usmar VM, Capaldi RA, Takamiya S et al: Reconstitution and molecular analysis of the respiratory chain; in Darley-Usmar VM, Rickwood D, Wilson MT (eds): Mitochondria a Practical Approach. Washington, DC, USA: IRL press, 1987, pp 113–152.
Muramoto K, Hirata K, Shinzawa-Itoh K et al: A histidine residue acting as a controlling site for dioxygen reduction and proton pumping by cytochrome c oxidase. Proc Natl Acad Sci USA 2007; 104: 7881–7886.
Kellogg EH, Leaver-Fay A, Baker D : Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins 2011; 79: 830–838.
Soiferman D, Ayalon O, Weissman S, Saada A : The effect of small molecules on nuclear-encoded translation diseases. Biochimie 2014; 100: 184–191.
Golubitzky A, Dan P, Weissman S, Link G, Wikstrom JD, Saada A : Screening for active small molecules in mitochondrial complex I deficient patient's fibroblasts, reveals AICAR as the most beneficial compound. PLoS One 2011; 6: e26883.
Gurgel-Giannetti J, Oliveira G, Brasileiro Filho G, Martins P, Vainzof M, Hirano M : Mitochondrial cardioencephalomyopathy due to a novel SCO2 mutation in a Brazilian patient: case report and literature review. JAMA Neurol 2013; 70: 258–261.
Zeharia A, Shaag A, Pappo O et al: Acute infantile liver failure due to mutations in the TRMU gene. Am J Hum Genet 2009; 85: 401–407.
Horvath R, Kemp JP, Tuppen HA et al: Molecular basis of infantile reversible cytochrome c oxidase deficiency myopathy. Brain 2009; 132: 3165–3174.
Martinelli D, Catteruccia M, Piemonte F et al: EPI-743 reverses the progression of the pediatric mitochondrial disease—genetically defined Leigh Syndrome. Mol Genet Metab 2012; 107: 383–388.
Salviati L, Hernandez-Rosa E, Walker WF et al: Copper supplementation restores cytochrome c oxidase activity in cultured cells from patients with SCO2 mutations. Biochem J 2002; 363: 321–327.
Casarin A, Giorgi G, Pertegato V et al: Copper and bezafibrate cooperate to rescue cytochrome c oxidase deficiency in cells of patients with SCO2 mutations. Orphanet J Rare Dis 2012; 19: 21.
Boczonadi V, Smith PM, Pyle A et al: Altered 2-thiouridylation impairs mitochondrial translation in reversible infantile respiratory chain deficiency. Hum Mol Genet 2013; 22: 4602–4615.
Sharma P, Mongan PD : Ascorbate reduces superoxide production and improves mitochondrial respiratory chain function in human fibroblasts with electron transport chain deficiencies. Mitochondrion 2001; 1: 191–198.
Ohlenbusch A, Edvardson S, Skorpen J et al: Leukoencephalopathy with accumulated succinate is indicative of SDHAF1 related complex II deficiency. Orphanet J Rare Dis 2012; 7: 69.
Acknowledgements
Corinne Belaiche is acknowledged for excellent technical assistance. This work was financed in part by E-Rare grant GenoMit (SE) and by the Manackerman Charitable Trust Fund UK (to DS and AS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Abdulhag, U., Soiferman, D., Schueler-Furman, O. et al. Mitochondrial complex IV deficiency, caused by mutated COX6B1, is associated with encephalomyopathy, hydrocephalus and cardiomyopathy. Eur J Hum Genet 23, 159–164 (2015). https://doi.org/10.1038/ejhg.2014.85
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.85
This article is cited by
-
The transcription regulator ATF4 is a mediator of skeletal muscle aging
GeroScience (2023)
-
Nicotinamide riboside kinase-2 regulates metabolic adaptation in the ischemic heart
Journal of Molecular Medicine (2023)
-
Characterization of terminal-ileal and colonic Crohn’s disease in treatment-naïve paediatric patients based on transcriptomic profile using logistic regression
Journal of Translational Medicine (2021)
-
Identification of key pathways and genes in polycystic ovary syndrome via integrated bioinformatics analysis and prediction of small therapeutic molecules
Reproductive Biology and Endocrinology (2021)
-
Mitochondrial complex IV deficiency caused by a novel frameshift variant in MT-CO2 associated with myopathy and perturbed acylcarnitine profile
European Journal of Human Genetics (2019)
