
Abstract
We performed a genome-wide association study (GWAS) to identify the genes responsible for age-related hearing impairment (ARHI), the most common form of hearing impairment in the elderly. Analysis of common variants, with and without adjustment for stratification and environmental covariates, rare variants and interactions, as well as gene-set enrichment analysis, showed no variants with genome-wide significance. No evidence for replication of any previously reported genes was found. A study of the genetic architecture indicates for the first time that ARHI is highly polygenic in nature, with probably no major genes involved. The phenotype depends on the aggregated effect of a large number of SNPs, of which the individual effects are undetectable in a modestly powered GWAS. We estimated that 22% of the variance in our data set can be explained by the collective effect of all genotyped SNPs. A score analysis showed a modest enrichment in causative SNPs among the SNPs with a P-value below 0.01.
Similar content being viewed by others
INTRODUCTION
Age-related hearing loss is characterized by a symmetric sensorineural hearing loss that is most pronounced in the high frequencies. Age of onset, progression, and severity of age-related hearing impairment (ARHI) show great variation in the population, but with a demonstrable increased prevalence in males.1 Heritability studies have shown that the sources of this variance are both genetic and environmental, with approximately half of the variance attributable to hereditary factors.2
Only a limited number of genome-wide association studies (GWASs) have been performed up to now. The first GWAS into ARHI reported an association between ARHI and SNPs within the GRM7 gene. This gene encodes the metabotropic glutamate receptor type7, which is activated through L-glutamate, the primary excitatory neurotransmitter in the cochlear hair cell.3 A GWAS in 352 samples from the Saami, an isolated population originating from northern Finland, revealed no genome-wide significant associations for ARHI, although the authors noted one SNP immediately downstream of the GRM7 gene among the most significant association signals.4 In a study on normal hearing function in 3417 persons over age 18, Girotto et al5 performed a GWAS in 6 isolated European populations using a meta-analysis approach. No SNPs were identified with genome-wide significance, but one of the most significant SNPs was found in the GRM8 gene, a close homolog of the GRM7 gene that contained several significant SNPs in previous association studies.
Large-scale studies on the genetic architecture of complex diseases and traits have indicated that these traits are usually polygenic in nature: their phenotypic variance is influenced by many genetic variants, each which of only contributes a very small fraction of the variance.
Studies on the genetic architecture of adult height, BMI, and psychiatric diseases indicate that even SNPs that fail to reach genome-wide significance substantially contribute to the phenotypic variance. Although it is not possible to pinpoint these SNPs individually, their collective effect can be estimated, and they may be one of the factors involved in the paradox of the missing heritability.6, 7, 8
As part of our continuing effort to elucidate the genetic and environmental causes of ARHI, we have enlarged our sample set to 2161 individuals all originating from Antwerp. Here, we present the results of a GWAS on ARHI, with and without adjustment for environmental risk factors, compare our results with previous studies, and look for potential pathways using pathway analysis. Apart from the classic association tests on common variants, we also look for the effect of rare variants and the interactions between SNPs on ARHI. Furthermore, we discuss the implications for the genetic architecture of ARHI and compare this with the findings for other complex traits.
MATERIALS AND METHODS
Sample collection and phenotyping
Subjects were collected at the audiological center from the Antwerp University Hospital using the selection criteria and questionnaire we described previously.9 Phenotypes were described using Principal component (PC) scores, as described in Supplementary data. In total, PC scores were obtained for 2161 individuals.
Sample selection for the three PC phenotypes
Each PC score has an approximately normal distribution. Power calculations (not shown) showed that most power for the subsequent association analyses resides within the samples with a relatively large or small PC score. Specifically, the most informative samples consist of those within the 20% highest and 20% lowest PC scores. Selecting only these samples reduced the genotyping effort, with little effect on the total power.
To make a sample selection based upon the three PCs simultaneously, maintaining high power for associations tests on all three PCs, sample selection was carried out in two steps, as shown in Figure 1: First, we flagged the samples that were within the 20% highest or lowest percentile of the distribution for each of the three PCs separately. The final selection for genotyping was obtained by including the samples that were flagged for at least one of the three PCs. Hence, samples that were never flagged in any of the three PCs, representing samples that had low information content for any of the three PCs, were not selected.

Schematic representation of the sample selection. Starting from a total sample size of 2161, the most informative samples were selected for genotyping based upon principle component analysis. Samples with an extreme value for either one of the first three PCs were included for genotyping. After various quality control steps, 1489 samples were included in the association analyses.
Genotyping
DNA was extracted from blood using standard procedures.
Genomic DNA concentrations were determined with PicoGreen (Invitrogen, Carlsbad, CA, USA). Additionally, the quality of the genomic DNA was assessed for each sample by gel electrophoresis. Five hundred DNA samples were genotyped with the Illumina CNV370 quad chip, 1060 DNA samples using the Illumina HumanOmniExpress BeadChip (Illumina, Inc., San Diego, CA, USA).
Imputation and filtering
Pre-imputation filtering was carried out to exclude SNPs with a minor allele frequency (MAF) below 1%, a P-value for Hardy–Weinberg equilibrium below 10e−6, a call rate below 95% across all samples and across all SNPs. Duplicate samples were identified using pi-hat analysis (pi-hat value>0.99). For each of these duplicates, we removed the sample with the lowest genotyping call rate.
Imputation was performed using the program impute2, version 2.1.2,10 with the 1000 Genomes Phase 1 Interim panel (June 2011) as a reference panel. Imputation resulted in a total of 11 626 570 SNPs. An additional post-imputation filtering was carried out using the same exclusion criteria as in the pre-imputation stage, and removing imputed SNPs with an Impute2 info metric below 0.5.
Association analysis
The association between the common variant genotypes (MAF>0.01) and the scores of the first three PCs was analyzed using the linear regression option in the software package PLINK v1.07 (http://pngu.mgh.harvard.edu/purcell/plink/). An additive model was fitted taking the PC scores as a quantitative outcome variable. Significance of the association was tested through the Wald test. This association test was repeated using the Efficient Mixed-Model Association eXpedited (EMMAX) software (http://genetics.cs.ucla.edu/emmax/), to account for possible cryptic relatedness or population stratification. A cutoff for cryptic relatedness of 0.05 was used.
The association test accounting for environmental risk factors was performed using a linear regression model as implemented in PLINK, with the following covariates: exposure to occupational noise (binary variable), smoking (in pack years), solvents (binary variable) and alcohol (binary variables), and the BMI. We refer to our previous paper on environmental risk factors for ARHI for a detailed description on how these risk factors were ascertained and coded.11
Details on the analysis of rare variants, gene–gene interactions and pathway analysis are given in the Supplementary Methods. In all analyses except the gene–gene interaction analysis, the hearing phenotypes were treated as a quantitative trait. Although the sample selection was performed based upon informativity for the three separate PC phenotypes (Figure 1), subsequent association testing, rare variant analyses, genome-wide complex trait analysis (GCTA), and pathway analyses were performed on all included samples (N=1489), regardless what PC phenotype they were selected for. This means that an individual that was highly informative for PC1 was also included in the analyses on PC2 and PC3 and vice versa.
Genome-wide complex trait analysis
The estimate of the genetic variance was performed using the Genome-wide Complex Trait Analysis tool.12 Details on the variance components analysis are given in the Supplementary Methods.
The score analysis to test for polygenic inheritance of ARHI was carried out as follows: first, we randomly partitioned the data set into 10 equal subsets. Nine of the subsets were combined to build a discovery set and one subset was retained as a validation set (Discovery set: 1326 individuals, validation set: 163 individuals). In the discovery set, the P-values for association with PC1 were recalculated using PLINK in the same way as the original analysis. We then estimated the random effects of the SNPs using the Best Linear Unbiased Prediction (BLUP) method, as implemented in GCTA. This method estimates the effects of the individual SNPs on the phenotype. Using these effects, a phenotypic score was obtained for the individuals in the validation set using the score function in PLINK. This step was carried out using various subsets of SNPs, using different P-value cutoffs. The correlation between this phenotypic score and the observed PC1 score in the validation set was calculated. As a negative control, the correlation between the phenotypic score and an independent phenotype (the PC2 score) was calculated. This procedure was repeated 10 times, whereby each of the 10 subsets served exactly once as a validation set. For each P-value cutoff, the mean and standard error of the 10 correlation coefficients was calculated.
Public data access
Data from this study are publicly available through the following link https://tgen.org/research/research-divisions/neurogenomics/supplementary-data/gwas_polygenic_arhi_fransen_et_al.aspx.
RESULTS
Sample selection and genotyping
To study the genetic factors influencing the normal variation in ARHI, we collected a population-based sample of 2161 unrelated individuals from a residential area near Antwerp, Belgium. The sample was obtained through population registries made available by the local city councils, and not specifically enriched for hearing-impaired people. To make the population ethnically homogenous, we requested that at least three out of the four grandparents originated from the same region as the study subject. No inbreeding was present in this population, and if full sibs were present, then only one of them was included in the study.
PC analysis was performed on the pure-tone audiometric data, adjusting for age and gender. In line with previous studies,4 we found that the first three PCs contain most of the information in the audiogram, with the PC1 score giving an overall measure of hearing capabilities across all frequencies, PC2 indicating whether the audiogram is flat or sloping, and PC3 being an indicator of the convexity/concavity of the audiogram. As shown in Figure 1, the 1560 most informative samples were selected for genotyping, based upon the PC scores for the first 3 PCs. Genotyping was performed using either the Illumina CNV370 quad chip or the Illumina HumanOmniExpress BeadChip, and the results from these two platforms were integrated using the program Impute2. After various quality control steps, a total of 4 167 292 markers remained for the association test in 1489 samples.
Common variant analysis
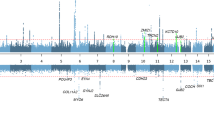
All SNPs that passed quality control were tested for association with the three first PCs. A list with all SNPs with a P-value below 1.0e−3 is given in Supplementary Table 1. Figure 2 shows a Manhattan plot, with the most significant genes highlighted in red. None of the markers included in our analysis reached the threshold for genome-wide significance. The most significant P-values were found for the association between the PC2 scores and several SNPs within the ACVR1B gene (minimum P-value=4.6e−7), and between the PC3 scores and an SNP in the CCBE1 gene (P=3.5e−7). For PC1, the most significant SNPs were an order of magnitude less significant, with a minimal P-value of 7.4e−6 for an SNP within the ZNF536 gene. None of the top-ranked SNPs in our study were statistically significant in any of the previous GWASs on ARHI.4, 5

Manhattan plot for the common variant analysis. The figure shows the logarithm of the P-values for the association of the genotypes versus each of the three PC phenotypes. P-values above 0.01 are not shown. Red dots indicate the SNPs in the genes that show the strongest association in the current study. Blue dots highlight the genes with a previously reported association with ARHI by Girotto et al,5 plus the GRM7 gene reported by Friedman et al3 and the IQGAP2 gene reported by Van Laer et al.4
To look for evidence of replication of previous studies, we first looked up the P-values for all individual SNPs that were reported to be significant in the previous GWASs and checked their significance in the current study. For the GWAS by Girotto et al,5 this included the SNPs having a P-value below 10e−5. For the GWAS on the Saami, we checked all SNPs with a P-value below 1.0e−4. For the study by Friedman et al,3 we selected the SNPs that were significant in the GRM7 gene and compared these with our PC1 results. The results are shown in Supplementary Table 2. In PC2, the current study found marginally significant associations for SNPs in the GPATCH3 and OTX2 genes. No significant P-values were found among the SNPs previously found significant in PC1 and PC3.
In Figure 2, blue dots highlight the SNPs in genes previously associated with ARHI, and reaching a P-value below 0.01 in the current study. The most remarkable gene is GRM8, with several SNPs reaching a P-value near 1e−4 for association with the PC1 score. None of the SNPs in the previously reported GRM7 gene showed a P-value below 0.001.
Common variants accounting for environmental risk factors
We and others previously showed that ARHI is significantly associated with several environmental risk factors, including occupational noise, smoking, high BMI, solvents, and alcohol.11 A detailed description on how these risk factors were ascertained can be found in this latter paper. We repeated the association test on the common variants taking into account exposure to the most significant environmental risk factors. The most significant associations for this analysis are graphically shown in Supplementary Figure 1. The ranking of the SNPs and genes differed compared with the analysis without covariate adjustment, but genome-wide significance was not reached (data not shown).
Common variants accounting for population stratification
To exclude an effect of cryptic relatedness between individuals or hidden population stratification on the test statistics, we compared the results from the common variant analysis with an association analysis using the software package EMMAX. This program implements a variance-component approach that accounts for empirically estimated relationships between the individuals when testing for association. As shown in Supplementary Table 1, the differences between the uncorrected association tests using PLINK and the corrected test in EMMAX are very small. The same genes and SNPs are found and show the strongest association, albeit with a slightly lower P-value in the adjusted analysis. The correlation between the P-values from the two methods equals 0.967, 0.999, and 0.994 for PC1, PC2, and PC3, respectively. λGC values for the uncorrected association analysis were 1.000, 0.995, and 1.002 for the first three PCs, whereas for the EMMAX analysis these values were 1.014, 0.997, and 1.001, respectively, confirming there was little effect of population stratification on the association results.
Rare variant analysis
Classic statistical tests offer low power to detect the effect of SNPs with low allele frequencies (MAF<0.05). To search for the effect of the rare variants on the phenotype, we used the Sequence Kernel Association Tests (SKAT).13 This test searches for the joint effects of multiple variants in a gene or a pre-defined region of the genome on a phenotype, without assuming that all rare variants have the same effect on the phenotype. We partitioned the genome into 13 000 regions according to the gene annotation, and performed the SKAT for every gene separately. Supplementary Table 3 lists the P-values obtained for each gene separately, for each of the three PCs. None of the genes reached the threshold for genome-wide significance (P=3.8e−6). The lowest observed P-value was 4.7e−5 for PC2.
Gene–gene interaction analysis
To test for the interaction between SNPs in different genes, we carried out the Gametic Phase Disequilibrium test as implemented in the software package SIXPAC.14 This analysis first filters candidate SNP pairs, by searching for strong LD between distant, physically unlinked SNPs in the cases alone. The shortlist of SNP pairs showing the strongest LD in cases is then followed up in the second step, which verifies whether the strength of the LD differs between cases and controls. This strategy identifies pairs of alleles from distant SNPs co-occurring more often in cases compared with controls, which could be indicative of gene–gene interactions. Since the search for high LD is not exhaustive, and the algorithm is non-deterministic, the program was run multiple times. Supplementary Table 4 shows the most significant across all runs. As 629 437 SNPs were included into this analysis, the number of tests equals (629 4372–629 437)/2, times four genetic models per SNP pair. This leads to a Bonferroni-corrected significance threshold of 6.3e−14. None of the SNP pairs reached this threshold in our data set. The most significant interaction, that shows up in several of the SIXPAC runs, reaches a P-value of 4.2e−11 for an interaction between SNP rs877674 and SNP rs6952893. This latter gene lies within an intron of the DYNC1I1 gene on Chr7, while the former is situated between the BAI2 and SPOCD1 genes on Chr1.
Genome-wide complex trait analysis
To study the genetic architecture of ARHI, we have analyzed the GWAS results using the Genome-wide Complex Analysis Tool (GCTA).12 Variance component analysis estimated the percentage of variance explained by the SNPs, obtained as the ratio of the genetic variance to the total variance at 22%. This analysis adjusts for incomplete LD between the SNPs in the GWAS and the causative SNPs, but assumes that the MAF between causative SNPs and GWAS SNPs is the same. We repeated the analysis with an adjustment that the causal SNPs could have a lower MAF compared with the GWAS SNPs, but the estimate of the variance explained by the SNPs remained almost the same (21%). Although this result does not formally prove that the causal SNPs have the same MAF spectrum as the GWAS SNPs, there is no evidence for the causal SNPs to be enriched in SNPs with lower allele frequency. Due to the relatively small sample size, the standard error of the estimate was large, and a more accurate estimate can be obtained using a larger sample size.
The association results do not seem to indicate the presence of a major ARHI gene. To analyze whether the model of polygenic inheritance, which was reported for several other complex traits, also applies to ARHI, we performed a score analysis. First, we randomly split the individuals from our data set into 10 equal parts. Nine of the ten parts formed the discovery set and one part was retained as a validation set. Second, we estimated the effects of all individual SNPs in the discovery set. Third, using these estimated SNP effects, we predicted a phenotype score for the individuals from the validation set. This latter step was carried out using several subsets of SNPs, based upon different P-value cutoffs. Finally, the Pearson’s correlation between this phenotypic score and the observed phenotype in the validation set is calculated. Ten-fold cross-validation was performed to obtain standard errors of the estimated correlation coefficients.
Figure 3 shows the mean correlation coefficients and their standard errors for various P-value cutoffs. When only the most significant fraction of the SNPs is used to calculate the phenotypic score (P<0.001), the correlation between the phenotypic score and the observed phenotype only reached a value of 0.07. However, when SNPs are included using more liberal P-value cutoffs, correlation coefficients increase to ∼0.10 (P<0.01). Cutoff values beyond 0.01 do not lead to a further increase in correlation, suggesting that the group of SNPs with P<0.01 harbors the majority of the causal SNPs. To test whether the effect of the SNPs is specific, we have also calculated the correlation between the phenotypic score and an independent phenotype (PC2). Across all P-value cutoffs, the correlation coefficients from the score analysis were significantly higher than the correlation coefficients with the independent phenotype (not shown).

Score analysis. Pearson correlation coefficients between the observed phenotype (PC1) and the predicted phenotype from the individuals in the validation set are shown in black. Correlation coefficients between these predicted phenotypes and an independent phenotype (PC2), which served as a negative control, are shown in red. Error bars denote the standard errors obtained by 10-fold cross-validation.
Pathway analysis
To investigate whether genes in specific biological processes or pathways were enriched in SNPs with low P-values, gene-set enrichment analysis was carried out using the MAGENTA package. Supplementary Table 5 shows the result of the gene-set enrichment analysis for the three PC scores, as well as for the analysis adjusting for environmental risk factors. Upon FDR correction, only two gene sets show a significant enrichment: The arachidonic acid secretion pathway (PC1, 95th percentile cutoff) and the transforming growth factor beta (TGFβ signaling pathway (PC3, 75th percentile cutoff). The Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) signaling pathway is significantly enriched in low P-values in the analysis of PC2 adjusted for environmental risk factors.
DISCUSSION
Traditionally, GWASs concentrate on the significance of common variants in a particular disease. Power calculations using the program QUANTO15 showed that the current study design holds 80% power to reach genome-wide significance for SNPs accounting for 2% of the phenotypic variance of the trait. Doubling the sample size would allow the detection of SNPs accounting for 1% of the variance. The inability of this and previous studies to find consistently replicated genes associated with ARHI reflects the fact that there are no major genes involved in ARHI. The most significant SNPs we obtained in our GWAS are probably enriched with causative SNPs, but at the moment these SNPs cannot be individually identified.
An estimate of the collective effect of all SNPs in our data set has shown that 22% of the variance can be explained by genetic differences. This is still much less than the heritability estimates based upon epidemiological studies, which typically reach a heritability of 40–50%. Our estimate covers the combined effects of the common SNPs, including the non-significant ones (see further), gene–gene interactions and rare variants, but does not cover the influence of CNVs, epigenetics or a possible overestimation of the heritability in the epidemiological studies. These latter three reasons are possible explanations for the observed heritability gap. Alternatively, the 22% may be an underestimate if the MAF spectrum of the causative SNPs is strongly shifted toward SNPs with a low MAF.7 We have repeated the estimate of the variance explained accounting for this possibility, but obtained an estimate very close to the initial analysis. Our estimates have a large standard error, and larger sample sizes will lead to a more accurate estimate.
This is the fourth genome-wide association analysis into ARHI. Apart from the analysis of common variants, we have studied the effect of rare variants, gene–gene interactions, and gene-set enrichment analysis. All previous studies reported significant or near-significant associations between the phenotype and SNPs in genes encoding GRM7 and GRM8. A recent study by Newman et al16 showed association of several audiometric traits with GRM7 haplotypes, although the initial findings could not be replicated. Others and we have suggested that these genes be the prime candidates for association with ARHI. The current study also finds several SNPs within these two genes with suggestive P-values, but lacking genome-wide significance. To find out whether our current results support a role for GRM7 and GRM8 in ARHI, we studied the relationship between the previously reported SNPs and the most significant GRM7 and GRM8 SNPs in the current study. For both genes, the SNPs with low P-values in the current study are located in a different region of the gene compared with the previously reported SNPs, and linkage disequilibrium with these latter SNPs is very weak (not shown). Conversely, none of the previously reported associated SNPs shows any trend toward association in the current study. Hence, for both genes the current and the previously reported association signals appear to be completely independent, and no region of the GRM7 or GRM8 gene was ever found to be associated in more than one study.
The genes encoding GRM7 and GRM8 contained 1285 and 1509 SNPs, respectively, and both genes span over 800 kB. Even though the SNPs within these two genes are not completely independent due to linkage disequilibrium, association testing of so many SNPs leads to an inflated type 1 error: due to testing multiple hypotheses, it becomes very likely to observe highly significant P-values even in the absence of any true association. As long as no adjustment is made for the number of SNPs tested within a gene, GRM7 and GRM8 are very likely to contain significant SNPs in any GWAS due to the sheer size of these genes. In Supplementary Figure 2, we have plotted the observed distribution of P-values for the GRM7 and GRM8 SNPs versus the expected distribution in the absence of any association. There seems to be no evidence that the SNPs in these genes are enriched in low P-values. The current study shows one GRM7 SNP and several GRM8 SNPs with a significant P-value, and it cannot be excluded that one or more of the signals in these genes represent genuine associations. Conversely, our results do not mean that all previous associations in GRM7 or GRM8 represent false positives. All gene regions and SNPs that were reported previously can still be confirmed by replication, but the current result does not add support to any of the previously reported associations in GRM7 or GRM8.
The current data set is powered to detect large effects and no SNPs reached genome-wide significance. Unlike Crohn’s disease or AMD, there is no indication for the presence of any major genes, not even among the genes involved in monogenic hearing loss. Among the post-GWAS analyses, only the pathway analysis showed three hits that remained significant after correction for multiple testing. We found a significant involvement of the arachidonic acid secretion pathway, the JAK/STAT and TGFβ signaling pathways. Arachidonic acid metabolites are found in virtually all cells and tissues, and are involved in the modulation of cell function. In the inflammation cascade, arachidonic acid is the precursor of prostaglandins (PGs) and leukotrienes (LTs). One possible link with hearing loss is the suggestion that salicylate-induced ototoxicity can be mediated through altered levels of PGs and LTs in the perilymph.17, 18 The TGFβ signaling pathway is involved in a wide range of cellular processes including cell growth, differentiation, apoptosis, and cellular homeostasis, in both developing and adult organisms. In the developing mouse embryo, TGFβ proteins are expressed in the cochlear epithelium of the inner ear.19 In the adult inner ear, TGFβ is activated during an immune response.20 The JAK/STAT signaling pathway is a major signal transduction pathway. Disruptions of this pathway have been implicated in immune deficiencies and cancers.18 No link to inner ear pathology or hearing loss was found.
At the moment, it is too early to draw firm conclusions on these latter findings. This study should be regarded as a first, exploratory step into the discovery of the responsible genes and pathways. Combination of these data with other GWAS, and the integration with results from expression studies or resequencing efforts can eventually lead to better insights into the pathophysiology of ARHI.
Several recent papers have evaluated the genetic architecture of complex traits. One of their main findings is that many complex traits are highly polygenic in nature, sometimes involving one major gene but often without any variants with a substantial effect size. The genetic variance is not solely attributable to the SNPs reaching genome-wide significance, but is also due to the small effects of many causal alleles that never reach genome-wide significance. The collective effect of these undetectable SNPs can be evaluated using the SNPs with weakly significant P-values as predictors for the phenotype in an independent data set, and estimating how strongly the predicted phenotype correlates with the observed phenotype. Here, we have performed such a score analysis on the ARHI data, and show for the first time that the genetic architecture of ARHI is similar to other polygenic complex traits: including less-significant SNPs considerably improves the prediction of the phenotype in an independent data set. The number of causative SNPs was enriched when considering the set of SNPs with a P-value below 0.01.
These findings are important in view of the discussion on the missing heritability. One of the explanations for the discrepancy between the heritability observed in epidemiological studies, and the combined variance explained by the associated SNPs, is that part of the missing heritability is contained within the SNPs that do not reach genome-wide significance. For human height, Yang et al7 showed that these SNPs might completely explain the missing heritability. Our study indicates that for ARHI, this is at least partly the case. The signal contained in the weakly significant SNPs (P<0.01) is weak but significant, and larger than the information contained in the SNPs with a more stringent P-value (P<0.001). More accurate estimates will be obtained if larger sample sizes become available. As the sample size increases, the subset of SNPs with low P-values gradually becomes more enriched with truly causative SNPs, and the collective effect of these SNPs on the phenotype will therefore increase.
An estimate of the collective effect of all SNPs in our data set showed that 22% of the variance explained in the phenotype are due to genetic factors. The most significant SNPs we obtained in our GWAS are probably enriched with causative SNPs, but at the moment these SNPs cannot be individually identified. Increasing the sample size will likely result in several replicated SNPs, but the majority of causative SNPs are probably not detectable using GWAS.
References
Gates GA, Mills JH : Presbycusis. Lancet 2005; 366: 1111–1120.
Huang Q, Tang J : Age-related hearing loss or presbycusis. Eur Arch Otorhinolaryngol 2010; 267: 1179–1191.
Friedman RA, Van Laer L, Huentelman MJ et al: GRM7 variants confer susceptibility to age-related hearing impairment. Hum Mol Genet 2009; 18: 785–796.
Van Laer L, Huyghe JR, Hannula S et al: A genome-wide association study for age-related hearing impairment in the Saami. Eur J Hum Genet 2010; 18: 685–693.
Girotto G, Pirastu N, Sorice R et al: Hearing function and thresholds: a genome-wide association study in European isolated populations identifies new loci and pathways. J Med Genet 2011; 48: 369–374.
Purcell SM, Wray NR, Stone JL et al: Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009; 460: 748–752.
Yang J, Benyamin B, McEvoy BP et al: Common SNPs explain a large proportion of the heritability for human height. Nat Genet 2010; 42: 565–U131.
Lango Allen H, Estrada K, Lettre G et al: Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 2010; 467: 832–838.
Van Laer L, Van Eyken E, Fransen E et al: The grainyhead like 2 gene (GRHL2), alias TFCP2L3, is associated with age-related hearing impairment. Hum Mol Genet 2008; 17: 159–169.
Marchini J, Howie B, Myers S, McVean G, Donnelly P : A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 2007; 39: 906–913.
Fransen E, Topsakal V, Hendrickx J-J et al: Occupational noise, smoking, and a high body mass index are risk factors for age-related hearing impairment and moderate alcohol consumption is protective: a European population-based multicenter study. J Assoc Res Otolaryngol 2008; 9: 264–276.
Yang J, Lee SH, Goddard ME, Visscher PM : GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 2011; 88: 76–82.
Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X : Rare-variant association testing for sequencing data with the Sequence Kernel Association Test. Am J Hum Genet 2011; 89: 82–93.
Prabhu S, Pe’er I : Ultrafast genome-wide scan for SNP-SNP interactions in common complex disease. Genome Res 2012; 22: 2230–2240.
Gauderman WJ, Morrison JM QUANTO 1.1: A computer program for power and sample size calculations for genetic-epidemiology studies http://hydrauscedu/gxe, 2006.
Newman DL, Fisher LM, Ohmen J et al: GRM7 variants associated with age-related hearing loss based on auditory perception. Hear Res 2012; 294: 125–132.
Jung TT, Park YM, Miller SK, Rozehnal S, Woo HY, Baer W : Effect of exogenous arachidonic acid metabolites applied on round window membrane on hearing and their levels in the perilymph. Acta Otolaryngol Suppl 1992; 493: 171–176.
Aaronson DS, Horvath CM : A road map for those who don’t know JAK-STAT. Science 2002; 296: 1653–1655.
Paradies NE, Sanford LP, Doetschman T, Friedman RA : Developmental expression of the TGF beta s in the mouse cochlea. Mech Dev 1998; 79: 165–168.
Satoh H, Billings P, Firestein GS, Harris JP, Keithley EM : Transforming growth factor beta expression during an inner ear immune response. Ann Otol Rhinol Laryngol 2006; 115: 81–88.
Acknowledgements
SB holds a doctoral fellowship of the FWO Vlaanderen. IS is a postdoctoral fellow of the FWO Vlaanderen. We wish to thank Jian Yang and Peter M Visscher (Queensland Institute of Medical Research) for help with the analysis using the GCTA software. This research was supported by funding from the Belgian Science Policy Office Interuniversity Attraction Poles (BELSPO-IAP) programme through the project IAP P7/43-BeMGI, through R01 grant DC010215, the Seaver Foundation, the Schwartz Foundation, and the State of Arizona.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Fransen, E., Bonneux, S., Corneveaux, J. et al. Genome-wide association analysis demonstrates the highly polygenic character of age-related hearing impairment. Eur J Hum Genet 23, 110–115 (2015). https://doi.org/10.1038/ejhg.2014.56
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.56
This article is cited by
-
Age-related hearing loss and its potential drug candidates: a systematic review
Chinese Medicine (2023)
-
Imputation of SNPs associated with presbycusis through linkage disequilibrium analysis in the ILDR1 gene
Journal of Genetics (2023)
-
Population-scale analysis of common and rare genetic variation associated with hearing loss in adults
Communications Biology (2022)
-
The genetic architecture of age-related hearing impairment revealed by genome-wide association analysis
Communications Biology (2021)
-
Genomewide association study of C-peptide surfaces key regulatory genes in Indians
Journal of Genetics (2019)

{kind=link}