
Abstract
Among the single-nucleotide polymorphisms (SNPs) previously reported to be associated with body mass index (BMI) and obesity, we focus on a common risk variant rs7566605 upstream of the insulin-induced gene 2 (INSIG2) gene and a rare protective variant rs2229616 on the melanocortin-4 receptor (MC4R) gene. INSIG2 is involved in adipogenesis and MC4R effects hormonal appetite control in response to the amount of adipose tissue. The influence of rs2229616 (MC4R) on BMI and obesity has been confirmed repeatedly and insight into the underlying mechanism provided. However, a main effect of rs7566605 (INSIG2) is under debate because of inconsistent replications of association. Interaction of rs7566605 with age may offer an explanation. SNP–age and SNP–SNP interaction models were tested on independent individuals from three population-based longitudinal cohorts, restricting the analysis to an observed age of 25–74 years. KORA S3/F3, KORA S4/F4 (Augsburg, Germany, 1994–2005, 1999–2008), and Framingham-Offspring data (Framingham, USA, 1971–2001) were analysed, with a total sample size of N=6926 in the joint analysis. The effect of interaction between rs7566605 and age on BMI and obesity status is significant and consistent across studies. This new evidence for rs7566605 (INSIG2) complements previous research. In addition, the interaction effect of rs7566605 with the MC4R variant rs2229616 on BMI was observed. This effect size was three times larger than that in a previously reported single-locus main effect of rs2229616. This leads to the conclusion that SNP–age or SNP–SNP interactions can mask genetic effects for complex diseases if left unaccounted for.
Similar content being viewed by others
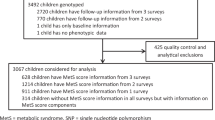
Introduction
Obesity is prevalent in the Western world, posing a serious health risk.1 Genes are known to contribute to its pathogenesis.2 The discovery of genetic interactions with age, other genes or the environment may improve consistency of associations as well as the understanding of the mechanisms leading to or underlying obesity. Here we present longitudinal interaction analyses for two candidate single-nucleotide polymorphisms (SNPs); one located upstream of the insulin-induced gene 2 (INSIG2) on chromosome 2 and the other in the melanocortin-4 receptor gene (MC4R) on chromosome 18. INSIG2 rs7566605 and MC4R rs2229616 were the first SNPs with a replicated polygenic effect on body mass in the general population3, 4 and known functional implications.5, 6
The INSIG2 region has been connected with obesity in human linkage studies7 as well as in one of the first genome-wide association studies (GWAS)3 to address obesity. This GWAS identified common variant rs7566605, located 10-kb upstream of the INSIG2 transcription start site. Elevated body mass index (BMI) or increased obesity risk was found for minor allele homozygotes (∼11% Caucasians) in at least six independent studies.3, 8 INSIG2 rs7566605 interaction analyses are of particular interest, as its main effect on BMI or obesity status was questioned following equal numbers of replications and non-replications of association in >20 well-powered data sets.3, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17 Combining all study results on INSIG2 suggest age as the most probable interaction candidate. A large meta-analysis18 (34 studies, 74 345 individuals) confirmed association of INSIG2 with obesity across study designs when comparing extremes (eg, BMI ≥32.5 kg/m2 versus BMI <25 kg/m2). No heterogeneity in INSIG2 main effect estimates was found between studies with higher compared with lower mean subject age; however, contrasted studies overlapped considerably in age range, with mean ages lying between 41 and 58 years.18 Nevertheless, this meta-analysis revealed an increased obesity risk for rs7566605 minor allele homozygotes in general population studies (odds ratio 1.092, P=0.035, 48 844 subjects from 16 studies), but a decreased obesity risk in population-based studies with subjects selected for better health status (odds ratio 0.796, P=0.028, 7640 subjects from 5 studies). Six other studies also indicated a tendency toward a protective effect of rs7566605 risk genotype CC on BMI or obesity status,3, 9, 10, 12, 14 or on waist-to-hip ratio.16 The effect strength of rs7566605 association varied between the first six longitudinal examinations on unrelated Framingham-Offspring subjects.8 Tests for rs7566605–age interaction were not significant for early childhood to middle age (4–50 years).19, 20 However, a lower gain in weight-for-length was reported for genotype CC babies between birth and the age of 6 months.21 These findings suggest an INSIG2–age interaction with weak marginal effect because of crossing BMI-age trends for different INSIG2 genotypes at a mid-life age (Figure 1, left). In this case, contrasting BMI extremes is robust but the heterogeneity test will not detect this, and power and sign of marginal effect estimates depend on the age range studied.22, 23

INSIG2–age interaction on longitudinal BMI. Left: growth curves of raw BMI for INSIG2 genotypes in men (joint analysis: FHS-Off, KORA3, KORA4). Displayed is a corridor of ±1 SD of the random subject-effect on intercept. Random subject-effects on the regression coefficient were small. Similar results were obtained for women and in analyses of rank-normalized BMI (Table 2). Right: INSIG2–age interaction on residual BMI (joint model: FHS-Off examinations 3 (1983–1987) and 5 (1991–1995); KORA3, KORA4). Displayed are the median and interquartile of residual BMI, after adjusting raw BMI for sex, baseline age, education and cohort. INSIG2 genotype is displayed by box colour (GG=white, GC=light grey, CC=dark grey), baseline age class by figure column. Longitudinal examinations T1 (KORA baseline, FHS-Off examination 3) and T2 (KORA follow-up, FHS-Off examination 5) are presented adjacent to each other.
INSIG2 fulfils a role complementary to insulin-induced gene 1 (INSIG1) in the regulation of cholesterol and triglyceride levels.24, 25 However, both genes also have separate additional functions. INSIG2 (but not INSIG1) is predominant in adipogenesis with a marked increase in gene expression during adipocyte differentiation.5 Multi-marker tag-SNP haplotypes in the close neighbourhood of rs7566605 associate with waist-to-hip ratio and computed tomography measures of visceral and subcutaneous adipose tissue.26 This suggests that rs7566605 tags a variant for altered adipogenesis.
This interaction analysis also examined the functional SNP rs2229616 in MC4R, which contributes to hormonal appetite control in response to the amount of adipose tissue.1, 27 MC4R harbours several susceptibility loci for obesity.6, 28, 29 Known functional SNPs in MC4R are all rare, have been found mostly in obesity studies and mostly associate with elevated BMI. The missense variant V103I (rs2229616), however, is relatively frequent and well-studied in population-based samples.4, 30, 31, 32, 33, 34 Minor allele carriers (∼3% Caucasians) have lower BMI,30, 31 lower risk of obesity4, 32, 33 and metabolic syndrome,34 and beneficially altered triglyceride and HDL-cholesterol levels.34, 35 Concurrently, functional studies demonstrated that V103I alters MC4R receptor responsiveness to its endogenous antagonist agouti-related protein.6
In this candidate study, we tested INSIG2 interaction with age and MC4R variant V103I on individual-level BMI data of unrelated adults (25–74 years) from three large longitudinal population-based cohorts. No other SNPs or hypotheses were tested.
Materials and methods
Samples
We analysed independent individuals from the KORA S3/F3 and KORA S4/F4 cohorts30, 36 (from the KORA study, http://www.helmholtz-muenchen.de/en/kora), and from the Framingham Heart Study Offspring cohort (FHS-Off)37 (Genetic Analysis Workshop 16 Framingham data, accession number phs000128.v1.p1 from the Database of Genotypes and Phenotypes (dbGaP), http://www.ncbi.nlm.nih.gov/gap). Data were retrieved and analysed in compliance with the Declaration of Helsinki. All studies were approved by local, regional and national ethics committees as required.
The KORA platform (Cooperative Research in the Region of Augsburg) recruited from the adult general population of the southern German city of Augsburg and its two adjacent districts.30, 36 The aim was to describe the prevalence of common diseases and their risk factors. We analysed baseline survey S3 (1994–1995) with 10-year follow-up F3 (KORA3), and baseline survey S4 (1999–2001) with 7-year follow-up F4 (KORA4).
Furthermore, we analysed independent subjects from the FHS-Off37 (26 years of follow-up: examinations 1 (1971–1975), 3 (1983–1987), 5 (1991–1995), 7 (1998–2001)). Subjects were adults recruited from the town of Framingham, MA, USA, with the identification of common factors in cardiovascular disease as objective. We excluded subjects on cholesterol treatment, as INSIG2 contributes to the regulation of cholesterol synthesis.
BMI is known to exhibit a gain phase up to late mid-life (55–62 years, dependent on study and sex); followed by diminished gain and a plateau or even slight decline at older age.38 We analysed subjects (Table 1) whose examinations occurred between 25 and 74 years of age, and at least half their examinations within the BMI gain phase (age ≤62 years). For FHS-Off with 26-year follow-up and four examinations, this restricted the selected baseline age range to 25–46 years (accounting for any discrepancy between actual age on examination and scheduled age). For KORA3 and KORA4 with 10 or 7-year follow-up and two examinations, this restricted the selected baseline age range to the whole adult BMI gain phase (25–62 years). The cohorts are otherwise comparable, including minor allele frequencies of considered SNPs and duration between examinations.
Statistical analysis
We performed longitudinal interaction analyses of rs7566605 with age and rs2229616 on BMI and obesity status. An additive model is not powerful for a recessive SNP (INSIG2)3 and its interaction with a dominant SNP (MC4R).4 We therefore used co-dominant SNP coding. All analyses were adjusted for sex, age, study and education (see Appendix for details). All P-values are nominal and two sided.
BMI is distributed right-skewed. Rank normalization of longitudinal BMI (joint Blom-transformation39 of all examinations; all cohorts in the joint analysis) removed this skewness but maintained the longitudinal rank correlation and order of the BMI data. Rank-normalized BMI was analysed using growth curve models.40 The resulting relative estimates of effect size were converted into absolute values (BMI units) by multiplying with the sample SD of BMI (KORA3: 4.5 kg/m2, FHS-Off, KORA4: 4.8 kg/m2, joint analysis: 4.7 kg/m2). Longitudinal obesity status (BMI ≥30 kg/m2) was analysed by logistic regression, accounting for dependence by random subject intercept or by solving generalized estimating equations with an unstructured working correlation matrix. The longitudinal nonparametric rank-sum test LNPT41, 42 validated the parametric analyses. LNPT resembles special repeated-measures ANOVA on rank order data42 of BMI, necessitating conversion of predictors and covariates into factors. Hence, baseline age is categorized for LNPT; cutoffs (Tables 2 and 3) ensure balanced classes for each cohort. LNPT was applied on raw BMI (adjusting for covariates within LNPT) and on residual BMI (after a priori covariate adjustment of raw BMI, see Appendix). FHS-Off and KORA cover the same age range. LNPT models the longitudinal study design by the factor longitudinal time course (examination number) and adjusts for baseline age class. However, power of the parametric models is optimized by using the covariable age on examination directly.42
Results
INSIG2 rs7566605–age interaction on BMI
We detected SNP–age interaction for INSIG2 rs7566605, but not for MC4R rs2229616. The BMI growth curve (Table 2, upper panel) of INSIG2 minor allele carriers has a lower intercept and significantly increased regression coefficient compared with the GG genotype (FHS-Off, KORA3, joint analysis). KORA3 and KORA4 contributed 46% and 43% of the total sample size to the joint analysis. Longitudinal logistic regression of obesity status (Table 2, bottom panel) confirmed a SNP-age effect. Expectedly, binary trait obesity is less powerful compared with quantitative BMI; however, evidence of SNP–age interaction is consistent between both phenotype models. The initially lowered obesity odd for genotype CC increases significantly with age.
Interestingly, estimates of INSIG2 CC genotype interaction with age were greater in the full KORA4 cohort (0.02 kg/m2 per year more than for GG genotype subjects, P=0.124, with negative intercept estimate), in contrast to the age-restricted KORA4 sample (Table 2), in which 835 subjects with baseline age >62 years were excluded. An INSIG2 main effects model also estimated greater CC genotype effect in the full KORA4 cohort (0.64 kg/m2, P=0.0015, in agreement with Herbert et al3) when compared with the age-restricted KORA4 sample (0.42 kg/m2, P=0.0656). The GC genotype’s main effect, however, was stable (full cohort: 0.29 kg/m2, P=0.0203; age-restricted sample: 0.32 kg/m2, P=0.0258; compared with the GG genotype). This strongly implies that INSIG2 genotype CC also interacts with age in KORA4.
SNP–age effect estimates were stable for different models of longitudinal correlation (comparing unstructured estimation of longitudinal covariance (data not shown) with parametric covariance models (Tables 2 and 3: autoregressive covariance (growth curve), or random subject intercept (logistic regression))). The interaction was also confirmed in raw BMI, albeit with weaker power (Supplementary Table). Further validation comes from the rank-sum test LNPT, which is model-free and free of distributional assumptions.42 LNPT on raw BMI (Table 2, middle panel, single cohorts) agreed strongly with LNPT on residuals of covariate-adjusted raw BMI (data not shown). LNPT tested two-way interactions of INSIG2 with baseline age (averaging over repeated examinations), or with longitudinal time course (averaging over the baseline age classes), and three-way interaction of INSIG2 with longitudinal time course and baseline age class. Power of the interaction tests with longitudinal time course42 is aided by the long-term follow-up (26 years) and relatively strong longitudinal autocorrelation of BMI (ρ=0.38) in FHS-Off. However, the two-way interaction test with baseline age is expected to have better power42 in KORA3 and KORA4, because of short-term follow-up (10, 7 years) and lower autocorrelation (ρ=0.16, 0.27). The three-way interaction tested for differential effects in older compared with younger subjects (which were suspected previously18). LNPT agreed with parametric results. INSIG2–age interaction on BMI was significant for FHS-Off (26 years follow-up, P=0.0079, three-way interaction with longitudinal time course and baseline age) and for KORA3 (10 years follow-up, P=0.0044, two-way interaction with baseline age) but not for KORA4. The three-way interaction in FHS-Off is due to a significant INSIG2 effect on longitudinal time course of BMI in older subjects (P=0.022, baseline age 34–46years), while no significance was reached in younger FHS-Off subjects (P=0.16, baseline age 25–33 years). LNPT joint analysis used FHS-Off examinations 3 and 5 to have balanced age classes (≤46 versus >46 years KORA baseline age, or FHS-Off age on examination 3) in each study. INSIG2 interaction with age on examination (Figure 1, left: growth curve) is consistent with INSIG2 interaction with longitudinal time course and age class (Figure 1, right: residuals of covariate-adjusted raw BMI, as analysed by LNPT). Growth curves of INSIG2 genotypes cross (Figure 1, left). Mean BMI of genotype CC subjects (bold dotted line) is initially lower compared with the other genotypes, but increases more rapidly with age and is elevated for ages ≥40 years. Consistently, LNPT detected no INSIG2 average effect but a tendency toward different marginal effects over the two baseline age classes (≤46 years/other; two-way interaction with age; joint analysis Table 2). P-values were also close to 0.05 for the interaction with examination (Table 2: joint analysis, comparable to KORA3).
MC4R–INSIG2 interaction on BMI
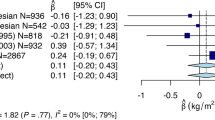
The final genetic interaction model contains the INSIG2–age interaction and tests for INSIG2–MC4R interaction effects on intercept (because of power considerations22). This model was tested in the large KORA cohorts and jointly over all three cohorts (ensuring >10 observations for all two-locus genotypes and all examinations). INSIG2–MC4R interaction was significant (P=0.001, Table 3, upper panel). Compared with additive SNP effects, we found an enhanced protective effect of the MC4R variant in INSIG2 heterozygotes (−1.26 kg/m2 on intercept (95% CI: −2.21, −0.31), P=0.009) and a tendency toward an enhanced detrimental effect in INSIG2 minor allele homozygotes who also have the MC4R variant (0.97 kg/m2 on intercept, P=0.148). A MC4R rs2229616 main effect was found in large, single-locus meta-analyses4, 30, 31, 32, 33 but was not detected by our interaction model. INSIG2 interaction with age was consistent between Tables 2 and 3. Growth curve results again were consistent with LNPT analyses of residuals of covariate-adjusted raw BMI (Table 3, Figure 2). However, LNPT had weaker power. LNPT P-values for the INSIG2–MC4R interaction were close to 0.05 whenever the growth curve model revealed significance (KORA3: P=0.036 interaction test for GC-GA subjects, excluding INSIG2 genotype CC, remaining subgroups have sufficient sample size; joint analysis: P=0.058 global test over all genotypes). Previous literature3, 4 on main effects reported that the INSIG2 SNP is recessive and the MC4R SNP is dominant. A recessive-dominant growth curve model of BMI confirmed the interaction of INSIG2 with MC4R and age (increased intercept of INSIG2–MC4R CC-GA genotype by 1.32 kg/m2 (95% CI: 0.10, 2.54), P=0.034; and increased regression coefficient for INSIG2 CC genotype by 0.02 kg/m2 per year, P=0.028).

MC4R–INSIG2 interaction on longitudinal BMI in population-based cohorts (joint model: FHS-Off examinations 3 (1983–1987) and 5 (1991–1995); KORA3, KORA4). Displayed are the median and interquartile of residual BMI, after adjusting raw BMI for sex, baseline age, education and cohort. INSIG2 rs7566605 genotype is displayed by box colour (GG=white, GC=light grey, CC=dark grey) in MC4R rs2229616 homozygotes (left) and heterozygotes (right); stratified by baseline age class (figure columns). Longitudinal examinations T1 (KORA baseline, FHS-Off examination 3) and T2 (KORA follow-up, FHS-Off examination 5) are displayed adjacent to each other. Sample sizes are given at the bottom of the graphs.
Discussion
Previous literature (Table 4) reported a recessive main effect3 of 0.60 kg/m2 for INSIG2 rs7566605 genotype CC in KORA4, with replication failures8 in FHS-Off and KORA3. In contrast, we provide consistent evidence of rs7566605–age interaction with both major statistical interaction models, testing interaction on both the additive scale (growth curve, LNPT) and multiplicative scale (logistic regression). MC4R rs2229616 was previously analysed for baseline examinations KORA S3 and S4, demonstrating a protective main effect30 of −0.52 kg/m2 for minor allele carriers. This may be a weaker marginal effect from a stronger INSIG2–MC4R interaction shown here (Table 3).
We found that INSIG2 minor allele homozygotes accumulate on average an excess of 1.86 kg/m2 in 62 years but have an MC4R genotype-dependent intercept (−0.05 kg/m2 for CC-GA genotype, −1.17 kg/m2 for CC-GG genotype), compared with GG-GG genotype subjects. The INSIG2–age effect is observed directly in INSIG2–MC4R CC-GA genotype subjects with a mean BMI elevated by 1.81 kg/m2 at the age of 62 years (∼0.4% Caucasians). In contrast, the mean BMI of CC-GG genotype subjects is lowered by −0.42 kg/m2 at the age of 25 years and raised by 0.69 kg/m2 at the age of 62 years (∼10.8% Caucasians). In contrast, mean BMI for GC-GA genotype subjects is lowered by −1.32 kg/m2 with no significant genotype–age effects (∼1.6% Caucasians). Hence, elderly INSIG2–MC4R CC-GA genotype subjects are particularly at risk of obesity. This also may explain previous INSIG2 main effect replication failures, suggesting that the proportion of younger and elderly subjects in a study can influence association outcome when not accounting for SNP–age interaction.
The strengths of this study are the modelling of individual participant data from high quality, well-characterized, population-based studies (no meta-analysis). The presented effects were robust with respect to different modelling approaches. In contrast to previous analyses, we restricted our analysis to subject age, motivated by the general dependence of BMI on age.38 No interaction was found earlier between INSIG2 rs7566605 and the GWAS identified common SNP rs17782313 near MC4R.17 This interaction analysis, however, examined the functional SNP6 rs2229616 in MC4R.
A few study limitations exist. As with every other joint analysis, we had to accommodate differences in study design (baseline age and follow-up duration) and different covariate information. Follow-up in KORA is relatively short and sample size for INSIG2–MC4R interaction analysis is still relatively small because of the rare rs2229616 variant. Our finding of INSIG2–age interaction (altered adipogenesis) and of INSIG2–MC4R interaction (interplay between adipogenesis and hormonal appetite control) would require validation by an independent study. We conclude that gene–gene or gene–age interaction may explain differences in genetic main effects across studies. More elaborate modelling could tackle this important question in future, large, well-characterized studies. Our results also encourage investigation into interactions between other obesity genes and with MC4R.
References
Wilborn C, Beckham J, Campbell B et al: Obesity: prevalence, theories, medical consequences, management, and research directions. J Int Soc Sports Nutr 2005; 2: 4–31.
Bell CG, Walley AJ, Froguel P : The genetics of human obesity. Nat Rev Genet 2005; 6: 221–234.
Herbert A, Gerry NP, McQueen MB et al: A common genetic variant is associated with adult and childhood obesity. Science 2006; 312: 279–283.
Geller F, Reichwald K, Dempfle A et al: Melanocartin-4 receptor gene variant I103 is negatively associated with obesity. Am J Hum Genet 2004; 74: 572–581.
Krapivner S, Popov S, Chernogubova E et al: Insulin-induced gene 2 involvement in human adipocyte metabolism and body weight regulation. J Clin Endocrinol Metab 2008; 93: 1995–2001.
Xiang Z, Litherland SA, Sorensen NB et al: Pharmacological characterization of 40 human melanocortin-4 receptor polymorphisms with the endogenous proopiomelanocortin-derived agonists and the agouti-related protein (AGRP) antagonist. Biochemistry 2006; 45: 7277–7288.
Deng HW, Deng H, Liu YJ et al: A genomewide linkage scan for quantitative-trait loci for obesity phenotypes. Am J Hum Genet 2002; 70: 1138–1151.
Lyon HN, Emilsson V, Hinney A et al: The association of a SNP upstream of INSIG2 with body mass index is reproduced in several but not all cohorts [electronic article]. PLoS Genet 2007; 3: e61.
Dina C, Meyre D, Samson C et al: Comment on ‘a common genetic variant is associated with adult and childhood obesity’. Science 2007; 315: 187b.
Loos RJF, Barroso I, O’Rahilly S, Wareham NJ : Comment on: a common genetic variant is associated with adult and childhood obesity. Science 2007; 315: 187c.
Rosskopf D, Bornhorst A, Rimmbach C et al: Comment on: a common genetic variant is associated with adult and childhood obesity. Science 2007; 315: 187d.
Boes E, Kollerits B, Heid IM et al: INSIG2 polymorphism is neither associated with BMI nor with phenotypes of lipoprotein metabolism. Obesity 2008; 16: 827–833.
Smith AJP, Cooper JA, Li LK, Humphries SE : INSIG2 gene polymorphism is not associated with obesity in Caucasian, Afro-Caribbean and Indian subjects. Int J Obes 2007; 31: 1753–1755.
Kumar J, Sunkishala RR, Karthikeyan G, Sengupta S : The common genetic variant upstream of INISG2 gene is not associated with obesity in Indian population. Clin Genet 2007; 71: 415–418.
Feng Y, Dong H, Xiang Q et al: Lack of association between rs7566605 and obesity in a Chinese population. Hum Genet 2006; 120: 743–745.
Bressler J, Fornage M, Hanis CL et al: The INSIG2 rs7566605 genetic variant does not play a major role in obesity in a sample of 24 722 individuals from four cohorts. BMC Med Genet 2009; 10: 56.
Andreasen CH, Mogensen MS, Borch-Johnsen K et al: Non-replication of genome-wide based associations between common variants in INSIG2 and PFKP and obesity in studies of 18 014 Danes. PLoS One 2008; 3: e2872.
Heid IM, Huth C, Loos RJF et al: Meta-analysis of the INSIG2 association with obesity including 74 345 individuals: does heterogeneity of estimates relate to study design? [electronic article]. PLoS Genet 2009; 5: e1000694.
Liu G, Zhu H, Dong Y, Podolsky RH, Treiber FA, Snieder H : Influence of common variants in FTO and near INSIG2 and MC4R on growth curves for adiposity in African- and European-American youth. Eur J Epidemiol 2011; 26: 463–473.
Fornage M, Papanicolaou G, Lewis CE, Boerwinkle E, Siscovick DS : Common INSIG2 polymorphisms are associated with age-related changes in body size and high-density lipoprotein cholesterol from young adulthood to middle age. Metabolism 2010; 59: 1084–1091.
Wu AC, Gillman MW, Taveras EM, Litonjua AA : INSIG2 is associated with lower gain in weight-for-length between birth and age 6 month. Clin Med Pediatrics 2009; 3: 33–37.
Gauderman JW, Macgregor S, Briollais L et al: Longitudinal data analysis in pedigree studies. Genet Epidemiol 2003; 25: S18–S28.
Lasky-Su J, Lyon HN, Emilsson V et al: On the replication of genetic associations: Timing can be everything!. Am J Hum Genet 2008; 82: 849–858.
Goldstein JL, DeBose-Boyd RA, Brown MS : Protein sensors for membrane sterols. Cell 2006; 124: 35–46.
Engelking LJ, Liang G, Hammer RE et al: Schoenheimer effect explained-feedback regulation of cholesterol synthesis in mice mediated by Insig proteins. J Clin Invest 2005; 115: 2489–2498.
Talbert ME, Langefeld CD, Ziegler JT, Haffner SM, Norris JM, Bowden DW : INSIG2 SNPs associated with obesity and glucose homeostasis traits in hispanics: the IRAS family study. Obesity 2009; 17: 1554–1562.
Friedman JM : Modern science versus the stigma of obesity. Nat Med 2004; 10: 563–569.
Dempfle A, Hinney A, Heinzel-Gutenbrunner M et al: Large quantitative effect of melanocortin-4 receptor gene mutations on body mass index. J Med Genet 2004; 41: 795–800.
Hinney A, Bettecken T, Tarnow P et al: Prevalence, spectrum, and functional characterization of melanocortin-4 receptor gene mutations in a representative population-based sample and obese adulds from Germany. J Clin Endocrinol Metab 2006; 91: 1761–1769.
Heid IM, Vollmert C, Hinney A et al: Association of the 103I MC4R allele with decreased body mass in 7 937 participants of two population based surveys. J Med Genet 2005; 42: e21.
Guo Y, Lanktree MB, Taylor KC et al: Gene-centric meta-analyses of 108 912 individuals confirm known body mass index loci and reveal three novel signals. Hum Mol Genet 2013; 22: 184–201.
Young EH, Wareham NJ, Farooqi S et al: The V103I polymorphism of the MC4R gene and obesity: population based studies and meta-analysis of 29 563 individuals. Int J Obes 2007; 31: 1437–1441.
Stutzmann F, Vatin V, Cauchi S et al: Non-synonymous polymorphisms in melanocortin-4 receptor protect against obesity: the two facets of a Janus obesity gene. Hum Mol Genet 2007; 16: 1837–1844.
Heid IM, Vollmert C, Kronenberg F et al: Association of the MC4R V103I polymorphism with the metabolic syndrome: the KORA study. Obesity 2008; 16: 369–376.
Brönner G, Sattler AM, Hinney A et al: The 103I variant of the melanocortin 4 receptor is associated with low serum triglyceride levels. J Clin Endocrinol Metab 2006; 91: 535–538.
Löwel H, Döring A, Schneider A, Heier M, Thorand B, Meisinger C : The MONICA Augsburg surveys—basis for prospective cohort studies. Gesundheitswesen 2005; 67Suppl 1: S13–S18.
Cupples LA, Heard-Costa N, Lee M, Atwood LD : Genetics analysis workshop 16 problem 2: the Framingham Heart Study data. BMC Proc 2009; 3 (Suppl 7): S3.
Strug L, Sun L, Corey M : The genetics of cross-sectional and longitudinal body mass index [electronic article]. BMC Genet 2003; 4Suppl I: S14.
Kraja AT, Corbett J, Ping A et al: Rheumatoid arthritis, item response theory, Blom transformation, and mixed models [electronic article]. BMC Proc 2007; 1Suppl 1: S116.
Laird NM, Ware JH : Random-effects models for longitudinal data. Biometrics 1982; 38: 963–974.
Malzahn D, Balavarca Y, Lozano JP, Bickeböller H : Tests for candidate-gene interaction for longitudinal quantitative traits measured in a large cohort. BMC Proc 2009; 3Suppl 7: S80.
Malzahn D, Schillert A, Müller M, Bickeböller H : The longitudinal nonparametric test as a new tool to explore gene-gene and gene-time effects in cohorts. Genet Epidemiol 2010; 34: 469–478.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG grant Klinische Forschergruppe (KFO) 241: TP5, BI 576/5-1), by the German Federal Ministry of Education and Research – the German National Genome Research Network NGFN (BMBF grants 01GR0464, 01GS0422, 01GS0837), by a NIH subcontract from the Children’s Hospital, Boston, MA, USA, (prime grant 1 R01 DK075787-01A1), and by the Munich Center of Health Sciences of the LMU. We would like to thank all study participants and investigators who contributed the phenotype and genotype data. The KORA research platform was initiated and financed by the Helmholtz Zentrum Munich (former GSF-National Research Centre for Environment and Health), which is funded by the German Federal Ministry of Education and Research, and of the State of Bavaria. The Framingham Heart Study (FHS) was conducted and is supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with Boston University (N01 HC25195). FHS data were obtained through dbGaP for Genetic Analysis Workshop 16, supported by NIH grant R01 GM031575 from the National Institute of General Medical Sciences. This article was not prepared in collaboration with FHS investigators and does not necessarily reflect the opinions or views of the FHS, Boston University, or the NHLBI. Furthermore, we would like to express our warmest thanks to Anke Hinney for her assistance with the manuscript.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Appendix
Appendix
STATISTICAL ANALYSIS
Parametric longitudinal models of BMI and obesity status
Rank-normalized BMI  of subject i at examination t
of subject i at examination t

depends on age on examination ageit with multivariate normal errors ɛit with zero mean and longitudinal covariance ∑. Unstructured estimation of ∑ from the data (data not shown) yielded similar estimates but weaker power compared with an autoregressive covariance model (growth curve, Tables 2 and 3). Between-subjects model equations 2 and 3


distinguish population-level estimates β for fixed effects of genes, confounders and individual random effects v (bivariate normally distributed with zero mean, unstructured covariance matrix). Study-specific differences were adjusted by five-level factor het5 constructed from the available information on education and study. Factor het5 distinguished FHS-Off (education not available) and two education strata in KORA3 and KORA4. Using three levels (FHS-Off, and two education strata in pooled KORA3, KORA4) yielded results identical to Tables 2 and 3 because of a nonsignificant study effect between age-restricted KORA cohorts (P=0.071). SNP–SNP interaction analysis extended the individual intercept model equation 2 by a MC4R main-effect and an age-independent MC4R–INSIG2 interaction. Obesity status was modelled analogous with a logistic link function for binary trait BMI ≥30 kg/m2, accounting for longitudinal dependence by random subject intercepts v0i (Table 2; maximum-likelihood solutions obtained with vli=0). These estimates were verified by solving generalized estimating equations with an unstructured working correlation matrix (data not shown).
Nonparametric longitudinal rank-sum test LNPT
LNPT compares BMI distributions F= at repeated examinations t for groups of subjects with genotype g and covariate a (presented as factors). LNPT resembles heteroscedastic repeated-measures ANOVA on mid-ranks of longitudinal BMI.42 LNPT is adjusted for all possible interactions between the included factors (SNP genotype, categorical covariates, examination number), to ensure reliable type-1 error control.42 Subjects with incomplete longitudinal BMI can be included. We applied LNPT on raw BMI for single studies, adjusting for covariates within LNPT by the factors sex, education and baseline age class. To better incorporate differences in the cohort designs, we also performed a priori covariate adjustment on raw BMI Yi with a longitudinal fixed effects model
at repeated examinations t for groups of subjects with genotype g and covariate a (presented as factors). LNPT resembles heteroscedastic repeated-measures ANOVA on mid-ranks of longitudinal BMI.42 LNPT is adjusted for all possible interactions between the included factors (SNP genotype, categorical covariates, examination number), to ensure reliable type-1 error control.42 Subjects with incomplete longitudinal BMI can be included. We applied LNPT on raw BMI for single studies, adjusting for covariates within LNPT by the factors sex, education and baseline age class. To better incorporate differences in the cohort designs, we also performed a priori covariate adjustment on raw BMI Yi with a longitudinal fixed effects model

Three-level factor het3 adjusted for FHS-Off and two education strata in pooled KORA3, KORA4 (given a nonsignificant study effect between age-restricted KORA cohorts). Residual BMI  was analysed by LNPT for genetic effects. Removal of a population trend with baseline age does not remove subgroup trends in residual BMI.
was analysed by LNPT for genetic effects. Removal of a population trend with baseline age does not remove subgroup trends in residual BMI.
Rights and permissions
About this article
Cite this article
Malzahn, D., Müller-Nurasyid, M., Heid, I. et al. Controversial association results for INSIG2 on body mass index may be explained by interactions with age and with MC4R. Eur J Hum Genet 22, 1217–1224 (2014). https://doi.org/10.1038/ejhg.2014.3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.3
Keywords
This article is cited by
-
A comprehensive review of genetic causes of obesity
World Journal of Pediatrics (2024)
-
Tumour biology of obesity-related cancers: understanding the molecular concept for better diagnosis and treatment
Tumor Biology (2016)
