
Abstract
Loss-of-function variants in ANKRD11 were identified as the cause of KBG syndrome, an autosomal dominant syndrome with specific dental, neurobehavioural, craniofacial and skeletal anomalies. We present the largest cohort of KBG syndrome cases confirmed by ANKRD11 variants reported so far, consisting of 20 patients from 13 families. Sixteen patients were molecularly diagnosed by Sanger sequencing of ANKRD11, one familial case and three sporadic patients were diagnosed through whole-exome sequencing and one patient was identified through genomewide array analysis. All patients were evaluated by a clinical geneticist. Detailed orofacial phenotyping, including orthodontic evaluation, intra-oral photographs and orthopantomograms, was performed in 10 patients and revealed besides the hallmark feature of macrodontia of central upper incisors, several additional dental anomalies as oligodontia, talon cusps and macrodontia of other teeth. Three-dimensional (3D) stereophotogrammetry was performed in 14 patients and 3D analysis of patients compared with controls showed consistent facial dysmorphisms comprising a bulbous nasal tip, upturned nose with a broad base and a round or triangular face. Many patients exhibited neurobehavioural problems, such as autism spectrum disorder or hyperactivity. One-third of patients presented with (conductive) hearing loss. Congenital heart defects, velopharyngeal insufficiency and hip anomalies were less frequent. On the basis of our observations, we recommend cardiac assessment in children and regular hearing tests in all individuals with a molecular diagnosis of KBG syndrome. As ANKRD11 is a relatively common gene in which sequence variants have been identified in individuals with neurodevelopmental disorders, it seems an important contributor to the aetiology of both sporadic and familial cases.
Similar content being viewed by others


Introduction
KBG syndrome (OMIM 158050) is an autosomal dominant syndrome first described in 1975, and is characterized by specific neurobehavioural, dental, craniofacial and skeletal anomalies and short stature.1 Macrodontia of the upper central incisors of the permanent teeth is a hallmark feature. We identified haploinsufficiency of ankyrin repeat domain-containing protein 11 gene (ANKRD11) as the cause of the 16q24 microdeletion syndrome, on the basis of overlapping microdeletions.2, 3 Subsequently, through whole-exome sequencing (WES), loss-of-function variants in ANKRD11 were identified to cause KBG syndrome.4
Thus far, microdeletions involving ANKRD11 have been reported in nine sporadic patients and two families.2, 3, 5, 6, 7, 8, 9, 10 As ANKRD11 variants appear relatively frequent in patients with developmental delay, we aimed to study the genotype and phenotype correlation in KBG syndrome caused by ANKRD11 abnormalities. We present a comprehensive overview of the clinical and molecular characteristics of 20 so far unreported patients from 13 families with ANKRD11 aberrations and review the current literature on ANKRD11 mutations and deletions. In addition to regular medical examinations, we performed two-dimensional (2D) and three-dimensional (3D) imaging to objectively analyse oral and facial phenotypes. On the basis of the results of our study, we discuss various implications for the clinical management of KBG syndrome.
Patients and methods
All patients were recruited through routine clinical genetic diagnostic services at the Department of Human Genetics of the Radboud University Medical Center or referred through our international network of collaborators. Informed consent was obtained for inclusion in the studies and the use of medical data and photographs according to local ethics agreements.
Clinical investigations
All patients were clinically examined by one or more clinical geneticist(s) and clinical photographs were taken with consent of the patients and/or their parents. Previous medical and dental records were requested, if necessary. 3D stereophotogrammetry was performed in patients 1B, 1C, 1D, 1E, 2, 3, 4, 5, 6, 7A, 7B, 7C, 8 and 9. A full dental and orofacial examination, an orthopantomogram (OPG) as well as intra- and extraoral photographs were made in patients 1B–E, 2–4, 6, 8 and 9. Four patients from family 1 and patients 2 and 3 were also examined by the Centre for Special Dentistry at the Radboud University Medical Center.
3D imaging methods
Apart from conventional clinical 2D photographs, 3D stereophotographs were acquired using a 3D camera (3dMDCranial System, 3dMD LLC, Atlanta, GA, USA). All 3D stereophotographs were taken in natural head position and habitual occlusion (as far as possible for this specific patient group). During image acquisition, patients were asked to relax their facial musculature and keep their eyes open.
The 3D images were analysed and compared with composite faces of age- and sex-matched unaffected Dutch controls to define which facial features are distinctive for KBG syndrome. The composite control faces consisted of a large number of controls from a specific age group and gender. In this way, an average face was generated for comparison with this specific patient group.
Molecular investigations
Peripheral blood DNA was used for sequence analysis of the coding regions of the ANKRD11 gene using standard Sanger sequencing according to routine diagnostic protocols (primers and PCR conditions are provided in the Supplementary Data).
Genomewide array analysis was performed before ANKRD11 analysis in all patients using different array platforms according to local protocols by the referring institutions.
Microarray analysis in patient 13 was performed using a CytoSure ISCA 8 × 60 K array (Oxford Gene Technology, Begbroke, Oxfordshire, UK) according to the manufacturer’s protocol and data were analysed using the CytoSure interpret software v4.3.2, genomebuild: GRCh37 (hg19).
In patient 7A and parents, WES was performed on an Illumina HiSeq2000TM platform (Illumina Inc, San Diego, CA, USA) after enrichment with the Agilent SureSelect XT Human All Exon 50 Mb kit (Agilent Technologies, Santa Clara, CA, USA). After read alignment with Burrows–Wheeler Transform (BWA)11 and variant calling with Genome Analysis Toolkit (GATK),12 the annotation was done by the Department of Human Genetics of the Radboud University Medical Center using an in-house developed pipeline.13 In patients 7A, 7B and 7C, the variant was confirmed by Sanger sequencing.
WES in patient 9 and parents was performed on an Illumina HiSeq 2500 platform (Illumina Inc) after enrichment with the Agilent SureSelect XT Human All Exon 50 Mb kit (Agilent Technologies). The Illumina data were processed with GATK12 v3.1.1 according to the best practice guidelines http://gatkforums.broadinstitute.org/discussion/3238/best-practices-for-variant-discovery-in-dnaseq. Briefly, we mapped the pairs with BWA-MEM19 v0.7.5a, marked duplicates, merged lanes, realigned indels. Base recalibration did not improve our results, so this step was skipped. Next, GATK Haplotypecaller was used to call SNPs and indels on all samples simultaneously. Variant effect predictions and annotation was added using snpEFF and dbNSFP.14 Detecting de novo variants was done with GATK’s phase-by-transmission and filtering the Mendelian violations on the de novo model and coverage >10 ×. The variant was confirmed by Sanger sequencing in a diagnostic setting.
WES in patients 10 and 11 was performed in a family-based trio approach using Illumina technology (Illumina Inc). The sequencing was performed at Oxford Gene Technology and sequencing data were returned and analysed using software supplied from OGT. The variant was confirmed by Sanger sequencing.
Results
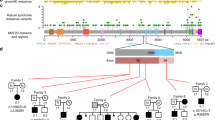
An overview of the clinical and molecular characteristics of families 1–12 is listed in Table 1 and Figure 1. All patients included in our study were clinically diagnosed with KBG syndrome compatible with the clinical criteria proposed by Skjei et al15 All our cases with normal array results were molecularly confirmed by the detection of heterozygous loss-of-function variants in ANKRD11. Table 2 shows the clinical features of patient 13 with a 1.16 Mb deletion encompassing exons 3–13 of the ANKRD11 gene (arr(hg19) 16q24.3(88231090–89388103) × 1) and an overview of the features of ANKRD11 microdeletion patients reported previously in other studies.2, 3, 5, 6, 7, 8, 9, 10 In total, we identified 11 different loss-of-function variants in ANKRD11 (Table 1; genomebuild GRCh37 (hg19), NM001256182.1).

Clinical features of families 1–11 with ANKRD11 mutations. a=1A, b=1B, c=1C, d=1D, e=1E, f=2, g=3, h=4, i=5, j=6, k=7A, l=7B, m=7C, n=8, o=9, p=10, q=11, r=12A, s=12B. The facial shape seems to evolve from round to more triangular at a later age, as seen in patient 5 (i), 7A (k) and 7C (m). All patients have an upturned nose with a broad base to the nose and full nasal tip. Other characteristic features are broad or bushy eyebrows with synophrys, strikingly prominent eyelashes (g, h, k, n, o), a low posterior hairline, brachy/turricephaly, a long philtrum, hypertelorism and prominent or protruding ears with dysplastic helices. Some patients have an exaggerated cupid’s bow-shaped mouth (a, i, k, m, n, o) but other patients have a thin upper lip (e, h, l, p, s). The hair can be coarse (a, b, d, l, q).
We performed segregation studies in all five affected members of family 1, three affected members of family 7 and in patient 12B. Carrier testing of the parents showed that the ANKRD11 mutations in patients 2, 3, 4, 6, 8, 9, 10 and 11 were de novo. The parents of patient 5 were not tested, but showed no clinical features of KBG syndrome.
Deposition of genetic data
The data obtained in this study are submitted to the LOVD (Leiden Open Variation Database), an online gene-centred collection and display of DNA variations (http://www.LOVD.nl/ANKRD11).
Dentofacial features
Macrodontia of upper permanent central incisors was present in all patients except patient 5, who was still in his transitional dentition. In patient 1D, macrodontia of the deciduous as well as the permanent dentition was observed as is shown in Figure 2. In patients 1B, 1C, 2, 3 and 9, macrodontia of other teeth, namely upper laterals and lower incisors, was noted as well. Hypodontia was seen in patients 1C and 1D and patient 3 (all of whom missed all four second premolars). Talon cusps were present in patients 1B, 1C and 2. Other dental anomalies were crowding (patient 1B), enamel hypoplasia (patient 9) and large dental pulps (patient 10). The dentofacial features of three patients of family 1, as well as patients 2, 3, 4, 6, 8 and 9 are shown in Figure 2. The dentofacial features of patient 13 with a 16q24 microdeletion are shown in Figure 3: the MRI image shows macrodontia of the permanent upper central incisors. The 2D clinical photograph (Figure 3) shows large upper central incisors in the deciduous dentition.

Clinical, intra-oral photographs and OPGs of three patients from family 1 (a–c), patient 4 (d), 6 (e), 2 (f), 8 (g), 9 (h) and 3 (i). All patients show macrodontia of upper central incisors. Patient 1D (a) shows macrodontia of central upper incisors and hypodontia of four permanent teeth. Patient 1B (b) shows macrodontia of four upper incisors, as well as talon cusps and dental crowding. Patient 1C (c) shows macrodontia of four upper incisors and hypodontia of four permanent teeth. Patients 4 and 6 (d and e) have no dental abnormalities except for macrodontia of upper central incisors (with a mesiodistal width ≥9.7 mm in both patients). Patient 2 (f) has macrodontia of the central upper incisors and talon cusps. Patient 8 (g) has rather large, mesially inclined central incisors and premature loss of the upper deciduous canines most probably due to crowding. Patient 9 (h) has macrodontia of upper central incisors and enamel defects. Dental anomalies of patient 3 (i) consist of macrodontia of four upper incisors and four lower incisors, as well as hypodontia of four premolars.

(a) Brain MRI of patient 13 with a 1.16 Mb microdeletion encompassing ANKRD11, showing macrodontia of the permanent dentition and lack of space between the teeth as a result of the macrodontia. (b) On clinical examination, the patient has facial features of KBG syndrome, including a short nose with a bulbous nasal tip, a round face, bushy eyebrows, prominent eyelashes, hypertelorism and an exagerrated cupid’s bow-shaped mouth (c) with large deciduous upper central incisors.
3D imaging
The analyses of 3D images of patients 5, 1D and 7C are shown in Figure 4. These two males and one female were considered representative for the KBG syndrome. In these three patients, the most striking shared facial feature is the bulbous nasal tip and the upturned nose with a broad base. Patient 5 has a more triangular-shaped face. In contrast, patient 1D has a round face. This is in concordance with our observation that the face seems to evolve from round at a young age to triangular shaped at a later age. Patient 7C has a relatively hypoplastic midface and chin compared with controls.

Two-dimensional clinical photographs and 3D stereophotogrammetry images of patients 5 (a), 1D (b) and 7C (c) are shown which were analysed and compared with composite faces of age- and sex-matched unaffected Dutch controls. Regions in green depict facial structures that are more prominent in the KBG syndrome patients. Red areas are more prominent in controls. In these three patients, the most striking shared facial feature is the bulbous nasal tip, which appears green, and the upturned nose with a broad base (which appears red because this part of the nose is hypoplastic when compared with controls). Patient 5 has a more triangular-shaped face, which is illustrated by the red areas on the lateral side of the face. In contrast, patient 1D has a round face (the lateral sides of the face are green). Patient 7C has a relatively hypoplastic midface and chin compared with controls. The full colour version of this figure is available at European Journal of Human Genetics online.
Discussion
Here, we present the largest cohort consisting of 20 patients with KBG syndrome molecularly confirmed by ANKRD11 aberrations so far, together with an overview of previously reported cases with either ANKRD11 mutations or 16q24 microdeletions encompassing ANKRD11. Besides the original six patients with ANKRD11, loss-of-function variants reported by Sirmaci et al,4 a de novo missense variant in ANKRD11 has been reported in an individual who also carried a 9q31.2-q33.1 microdeletion.16 Five patients with deletions encompassing solely the ANKRD11 gene have been described in the literature.3, 5, 8 In addition, eight other patients with larger deletions including several flanking genes (including ZNF778 and CDH15) have been reported.2, 7, 9, 10
ANKRD11 is expressed in the brain and localizes mainly to the nuclei of neurons and glial cells.4 ANKRD11 has two transcription repression domains located at the C- and N-terminals, respectively, and a transcription activating domain. The protein regulates ligand-dependent transcriptional activation through recruitment of histone deacetylases to the p160 coactivators/nuclear receptor complex.17 Furthermore, ANKRD11 was found to be a novel p53-interacting protein enhancing the transcriptional activity of p53, hence functioning as a p53 coactivator.18 All ANKRD11 variants identified in this study are clustering in exon 10, which is likely due to the fact that this is the largest exon (6577 base pairs in length). All variants identified are predicted to lead to a loss of function, which supports the hypothesis that haploinsuffiency of ANKRD11 causes KBG syndrome.4
Macrodontia of the permanent upper central incisors, defined as a mesiodistal width ≥10 mm in males and ≥9.7 mm in females, is present in all ANKRD11 mutation patients reported so far4, 16 and is present in all the cases in this study except for patient 5, but this patient was still in his transitional dentition. Macrodontia can also be present in the deciduous dentition of KBG syndrome patients, as is shown in patient 1D and 13, and this was also reported by the parents of patient 9. In addition, 5/20 patients (25%) exhibited macrodontia of additional teeth as well. Additional dental abnormalities that we observed included talon cusps, oligodontia, enamel defects, dental crowding and large dental pulps.
Retrospectively, all four originally described 16q24.3 microdeletion patients2 fulfilled the diagnostic criteria for KBG syndrome and their dental features were also consistent with KBG syndrome. On detailed orofacial examination by an orthodontic specialist, patient 1 of this previous study2 had rather large front teeth, but no macrodontia (C Carels, personal communication). He had a broad uvula with a dimple. Patient 4 of that study had macrodontia with fusion of the maxillary lateral and central incisors (E Gierkes, personal communication). These observations confirm the similarities in the dentofacial phenotype of ANKRD11 mutation and deletion patients.
Short stature is defined as a height below the 3rd centile or below two standard deviations (SDs). As we observed in patient 3 and 5, this may be the first presenting feature of KBG syndrome, and there is clinical overlap with other short stature syndromes such as Silver–Russell syndrome.10 In our cohort, 12/20 patients (60%) had short stature, ranging from −2 SD to −4 SD. Three patients presented with intrauterine growth retardation. Adult height was within the low-normal range (−0.5 to −2 SD) in 4/9 patients (44%), which shows that short stature does not always have to be present in KBG syndrome. Patient 11 was treated with growth hormone because of short stature and IGF1-deficiency. This treatment was successful, because his height increased from −2.5 SD to −0.75 SD.
With regards to the skeletal phenotype, all patients showed hand anomalies, mostly brachydactyly and/or clinodactyly of the 5th finger, as has been previously reported in the majority of KBG syndrome patients.15, 19 There were mild costovertebral anomalies in 7/20 patients. Few spinal radiograms were available for evaluation, and it is difficult to draw any conclusion if vertebral fusions and/or significant kyphohosis or scoliosis are common in KBG syndrome. A further systematic evaluation with regard to skeletal radiographic features is necessary to draw a conclusion regarding this. Patient 5 developed bilateral Perthes’ disease at the age of 5 years; patient 4 had unilateral Perthes’ disease at the age of 6 years and patient 7B had a congenital hip dysplasia. Hip dysplasia and short femoral necks have been reported before in KBG syndrome.19 This confirms the presence of hip anomalies, including Perthes’ disease, in molecularly confirmed KBG syndrome patients.
Hearing loss was previously reported in one patient with an ANKRD11 mutation4 and is present in 6/19 mutation patients (~32%) in our cohort. Patient 1C was diagnosed with hearing loss at the age of four. The age range of patients with hearing loss in our study is from 4 to 47 years. The hearing loss was conductive in all patients, except in patient 1C, who had a mixed hearing loss, but mostly conductive. There might be a causal relationship between the hearing loss and recurrent infections. As our data are not sufficient to draw this conclusion, more research is needed to investigate whether aggressive antibiotic treatment could prevent hearing loss in these patients. We recommend that children with ANKRD11 variants undergo regular hearing tests after diagnosis.
Congenital heart defects (CHDs) have been reported in three 16q24.3 deletion patients (Table 2) and in an Italian cohort of KBG syndrome patients.2, 7, 19 We show that three patients in our study have CHDs (15%) and thereby confirm that this is a minor feature of KBG syndrome caused by ANKRD11 variants, warranting cardiac examination after diagnosis in young children. Palatal abnormalities have been described in KBG patients and consist of a high-arched palate, submucous cleft palate, bifid uvula and/or velopharyngeal insufficiency (VPI).19, 20 In our cohort, patient 4 and patient 11 had palatal abnormalities, whereas patient 1D had hypernasal speech but no VPI. Interestingly, patient 4 has a history of VPI and a heart defect, and for years it was assumed that she had velo-cardio-facial (VCF) syndrome. This overlap in clinical features identifies KBG syndrome as a serious differential diagnostic alternative in individuals with VPI and developmental delay.
Mild-to-moderate ID is a key feature of KBG syndrome. However, patients may present with only minor learning difficulties. Patient 4 finished regular secondary education. Notably, two other patients in our cohort also have a normal IQ, with only a delay in speech development. Recently, a patient with a 16q24.3 microdeletion was reported, who also had a normal intelligence.10
With regard to epilepsy, five patients in our cohort suffered from generalized tonic–clonic seizures in their childhood, which responded well to anti-epileptic drugs. This is in concordance with earlier reports of seizures or absences occurring mostly in childhood and following a benign course.19, 21 Abnormalities on brain neuroimaging have been reported in one clinically diagnosed KBG syndrome patient22 and in two 16q24.3 deletion patients (with a deletion including ANKRD11, CDH15 and ZNF778) and one ANKRD11 deletion patient.9 Patient 9 has a pineal cyst, but no other abnormalities. Patient 10 showed slighty enlarged ventricles, and patient 11 showed dysgenesis of the corpus callosum. Brain MRI in patients 1B, 2 and 4 was normal. Brain MRI of patient 13 with a 16q24.3 microdeletion was normal except for mildly delayed myelination.
Almost all patients (18/20) in our cohort have behavioural abnormalities. In 11/20 (55%) patients, attention deficit hyperactivity disorder (ADHD) or hyperactive behaviour with or without autism spectrum disorder (ASD) is present. Hyperactive behaviour and anxious personality have been previously described in KBG patients4, 19, 21 with ADHD present in 28% of cases.21 Other behavioural abnormalities observed in our cohort are anxious personality, compulsive behaviour, aggressive behaviour and temper tantrums. So far, ASD had not been reported in KBG syndrome patients with ANKRD11 mutations.4, 21 However, it has been frequently noted in 16q24.3 microdeletion patients, questioning whether this feature might be attributable to haploinsufficiency of other genes flanking ANKRD11 in the 16q24.3 region.2, 3, 5, 6, 7, 8, 9, 21 As ASD is present in about half of our patients (47%) with ANKRD11 mutations, we believe that ASD is a part of the phenotypic spectrum of KBG syndrome caused by ANKRD11 haploinsufficiency.
When comparing all the features of the 14 deletion patients (Table 2) and 19 mutation patients (Table 1), the frequency of congenital anomalies, seizures and behavioural problems seems to be similar in both groups. Because of the small number of patients, it is difficult to assess whether the level of ID is correlated with the size of the deletion; in our study we did not observe such a clear correlation. However, a contribution of flanking genes to the severity of the phenotype cannot be excluded.
We have analysed the data of the patients in Tables 1 and 2 with regard to the criteria for a clinical diagnosis of KBG syndrome by Skjei et al15 (see Supplementary Data). According to these criteria, a clinical diagnosis of KBG syndrome is made when at least four of the major criteria are fulfilled. In the group of ANKRD11 mutation patients, all patients fulfilled four or more major criteria. In the deletion group, however, 38% of patients did not fulfil four or more major criteria. We noted that the criterion ‘significantly delayed bone age’ was only assessed in about half of all patients; therefore, only 15% of the total of 33 patients fulfilled this criterion. We propose that this criterion is not necessary for the diagnosis of KBG syndrome; three out of seven major criteria would be sufficient to clinically diagnose KBG syndrome.
This report further delineates the phenotype of KBG syndrome caused by ANKRD11 aberrations. On the basis of the clinical data of the patients in this study and the previously published data on patients with ANKRD11 aberrations, we conclude that the phenotypes of mutation and deletion patients are similar, but there is intra- and interfamilial variation in both patient groups. Our observations confirm that CHDs are a feature of KBG syndrome which is important for clinical management. Based on our clinical data and the literature we also recommend regular hearing tests for children with KBG syndrome since hearing loss is relatively common, can develop at a young age and thereby hamper speech and language development. KBG syndrome might well be underdiagnosed because of the usually mild cognitive deficits and sometimes subtle dysmorphic features. Therefore, one has to be aware that inherited or familial cases are common as was also observed in this study. With the increasing use of next-generation sequencing (NGS) to identify the cause of cognitive dysfunction, it is anticipated that more ANKRD11 variants will be reported in the near future. Accurate phenotyping of patients remains important, especially for the interpretation of these variants detected with NGS. The 3D imaging studies applied in three of our patients illustrate the distinctive facial phenotype of KBG syndrome and serve as a base for further 3D studies in our cohort of ANKRD11-positive patients. This will aid in the early diagnosis of KBG syndrome.
Change history
13 August 2015
This paper has been corrected since online publication and a corrigendum also appears in this issue.
References
Herrmann J, Pallister PD, Tiddy W, Opitz JM : The KBG syndrome – a syndrome of short stature, characteristic facies, mental retardation, macrodontia and skeletal anomalies. Birth Defects Orig Artic Ser 1975; 11: 7–18.
Willemsen MH, Fernandez BA, Bacino CA et al: Identification of ANKRD11 and ZNF778 as candidate genes for autism and variable cognitive impairment in the novel 16q24.3 microdeletion syndrome. Eur J Hum Genet 2010; 18: 429–435.
Isrie M, Hendriks Y, Gielissen N et al: Haploinsufficiency of ANKRD11 causes mild cognitive impairment, short stature and minor dysmorphisms. Eur J Hum Genet 2012; 20: 131–133.
Sirmaci A, Spiliopoulos M, Brancati F et al: Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am J Hum Genet 2011; 89: 289–294.
Youngs EL, Hellings JA, Butler MG : ANKRD11 deletion in a 17-year-old male. Clin Dysmorphol 2011; 20: 170–171.
Marshall CR, Noor A, Vincent JB et al: Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet 2008; 82: 477–488.
Sacharow S, Li D, Fan YS, Tekin M : Familial 16q24.3 microdeletion involving ANKRD11 causes a KBG-like syndrome. Am J Med Genet A 2012; 185A: 547–552.
Khalifa M, Stein J, Grau L et al: Partial deletion of ANKRD11 results in the KBG phenotype distinct from the 16q24.3 microdeletion syndrome. Am J Med Genet A 2013; 161A: 835–840.
Miyatake S, Murakami A, Okamoto N et al: A de novo deletion at 16q24.3 involving ANKRD11 in a Japanese patient with KBG syndrome. Am J Med Genet A 2013; 161A: 1073–1077.
Spengler S, Oehl-Jaschlowitz B, Begemann M et al: Haploinsufficiency of ANKRD11 (16q24.3) is not obligatory associated with cognitive impairment but shows a clinical overlap with Silver–Russel syndrome. Mol Syndromol 2013; 4: 246–249.
Li H, Durbin R : Fast and accurate short read alignment with Burrows–Wheeler Transform. Bioinformatics 2009; 25: 1754–1760.
McKenna A, Hanna M, Banks E et al: The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–1303.
De Ligt J, Willemsen MH, van Bon BW et al: Diagnostic exome sequencing in persons with severe intellectual disability. N Eng J Med 2012; 367: 1921–1929.
Cingolani P, Platts A, Wang le L et al: A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012; 6: 80–92.
Skjei KL, Martin MM, Slavotinek AM : KBG syndrome: report of twins, neurological characteristics, and delineation of diagnostic criteria. Am J Med Genet A 2007; 143A: 292–300.
Xu M, Zhou H, Yong J et al: A Chinese patient with KBG syndrome and a 9q31.2-33.1 microdeletion. Eur J Med Genet 2013; 56: 245–250.
Zhang A, Yeung PL, Li CW et al: Identification of a novel family of ankyrin repeats containing cofactors for p160 nuclear receptors coactivators. J Biol Chem 2004; 279: 33799–33805.
Neilsen PM, Cheney KM, Li CW et al: Identification of ANKRD11 as a p53 coactivator. J Cell Sci 2008; 121: 3541–3552.
Brancati F, D’Avanzo MG, Digilio MC et al: KBG syndrome in a cohort of Italian patients. Am J Med Genet 2004; 131A: 144–149.
Ockeloen CW, Simpson J, Urquhart J et al: Velopharyngeal insufficiency: high detection rate of genetic abnormalities if associated with additional features. Arch Dis Child 2013; 99: 52–57.
Lo-Castro A, Brancati F, Digilio MC et al: Neurobehavioral phenotype observed in KBG syndrome caused by ANKRD11 mutations. Am J Med Genet B Neuropsychiatr Genet 2013; 162B: 17–23.
Oegema R, Schot R, de Wit MC et al: KBG syndrome associated with periventricular nodular heterotopia. Clin Dysmorphol 2010; 19: 164–165.
Acknowledgements
We thank all our patients and their parents for their kind cooperation. We thank Gary McCullagh MRCPCH, Paediatric Neurologist in the Royal Manchester Childrens’s Hospital, for providing the MRI image of patient 13. This work was supported by grants from the The Netherlands Organization for Health Research and Development (ZonMw grant 907-00-365 to TK). EAJ was supported by the Manchester Biomedical Research Centre. GG was supported by the Foundation Promobilia, AN by Stiftelsen Frimurare Barnhuset i Stockholm and the Stockholm County Council (ALF project). BWMvB was supported by the Ter Meulen Fund.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Ockeloen, C., Willemsen, M., de Munnik, S. et al. Further delineation of the KBG syndrome phenotype caused by ANKRD11 aberrations. Eur J Hum Genet 23, 1176–1185 (2015). https://doi.org/10.1038/ejhg.2014.253
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.253
This article is cited by
-
DYSMORPHIC features and adult short stature: possible clinical markers of KBG syndrome
Italian Journal of Pediatrics (2021)
-
The effect of Ekman–Westborg and Julin trait (generalized megadontia) and localized megadontia on tooth components size and ion content
Clinical Oral Investigations (2021)
-
Haploinsufficiency for ANKRD11-flanking genes makes the difference between KBG and 16q24.3 microdeletion syndromes: 12 new cases
European Journal of Human Genetics (2017)
-
Molecular subtyping and improved treatment of neurodevelopmental disease
Genome Medicine (2016)
-
Long-term outdoor air pollution and DNA methylation in circulating monocytes: results from the Multi-Ethnic Study of Atherosclerosis (MESA)
Environmental Health (2016)
