
Abstract
Familial hypertrophic cardiomyopathy (HCM) is usually caused by autosomal dominant pathogenic mutations in genes encoding sarcomeric or sarcomere-associated cardiac muscle proteins. The disease mainly affects adults, although young children with severe HCM have also been reported. We describe four unrelated neonates with lethal cardiomyopathy, and performed molecular studies to identify the genetic defect. We also present a literature overview of reported patients with compound heterozygous or homozygous pathogenic MYBPC3 mutations and describe their clinical characteristics. All four children presented with feeding difficulties, failure to thrive, and dyspnea. They died from cardiac failure before age 13 weeks. Features of left ventricular noncompaction were diagnosed in three patients. In the fourth, hypertrabeculation was not a clear feature, but could not be excluded. All of them had septal defects. Two patients were compound heterozygotes for the pathogenic c.2373dup p.(Trp792fs) and c.2827C>T p.(Arg943*) mutations, and two were homozygous for the c.2373dup and c.2827C>T mutations. All patients with biallelic truncating pathogenic mutations in MYBPC3 reported so far (n=21) were diagnosed with severe cardiomyopathy and/or died within the first few months of life. In 62% (13/21), septal defects or a patent ductus arteriosus accompanied cardiomyopathy. In contrast to heterozygous pathogenic mutations, homozygous or compound heterozygous truncating pathogenic MYBPC3 mutations cause severe neonatal cardiomyopathy with features of left ventricular noncompaction and septal defects in approximately 60% of patients.
Similar content being viewed by others


Introduction
Hypertrophic cardiomyopathy (HCM) is a major cause of sudden cardiac death in people younger than age 35 years under physical stress, and a major cause of mortality and morbidity in the elderly. It has an estimated prevalence of 1 in 500 individuals. In ~60% of cases, a pathogenic variant in one of the sarcomeric contractile protein genes is found.1,2
Familial HCM is usually an autosomal dominant disorder that is transmitted due to heterozygous pathogenic gene mutations with incomplete penetrance. Variants in genes encoding proteins involved in the sarcomere, cytoskeleton and Z-disk, in calcium handling, and in mitochondrial and lysosomal functions have been associated with HCM.3 To date, >1000 different pathogenic mutations have been found in genes that encode sarcomeric proteins, such as β-cardiac myosin heavy chain (MYH7) and cardiac myosin-binding protein-C (MYBPC3).4
Interfamilial and intrafamilial clinical variability in HCM is high, and it is difficult to establish genotype–phenotype correlations.4 In addition to the primary genetic defect, the effects of modifier genes or additional sarcomeric gene variants may contribute to the phenotypic expression of HCM.5 Childhood-onset cardiac hypertrophy is also genetically determined in the majority of cases, and two-thirds of familial cases of childhood-onset cardiac hypertrophy are caused by a pathogenic mutation in one of the sarcomeric protein genes.6
We describe four unrelated children with severe cardiomyopathy, features of ventricular noncompaction, and septal defects due to compound heterozygosity or homozygosity for truncating pathogenic mutations in MYBPC3, resulting in early death.
Materials and methods
Clinical diagnosis
Four unrelated families with an index patient with severe neonatal cardiomyopathy were studied, after obtaining informed consent. Clinical evaluation included clinical history and physical examination, electrocardiography (ECG) and 2D and M-mode echocardiography. The clinical diagnosis of HCM is made when there is a hypertrophied (often asymmetric), non-dilated left ventricle on echocardiography (LVW≥15 mm) in the absence of other cardiac or systemic diseases.1 In children, diagnosis is made on the basis of left ventricular wall thickness two or more standard deviations above the normal population mean for body surface area.7 Noncompaction of the left ventricle was diagnosed based on three echocardiographic criteria defined by Jenni et al,8 including (1) a thick noncompacted (NC) endocardial layer in the end systole on parasternal short-axis views (ratio NC/C>2) with numerous, excessively prominent trabeculations and deep intertrabecular recesses, (2) these recesses were perfused on color Doppler studies, and (3) a predominantly apical localization.
Pathologic studies
Microscopic examination and electron microscopy of cardiac septal autopsy material of patient 1 were performed with standard techniques (hematoxylin and eosin staining, magnification × 100).
Molecular analysis
Genomic DNA of the patients was isolated from blood samples or fibroblasts. All coding regions and intron–exon boundaries of the MYBPC3 gene were analyzed by direct sequencing analysis as described.9 In addition, MLPA analysis of the MYBPC3 gene was performed (SALSA MLPA kit P100, MRC Holland, Amsterdam, the Netherlands) to detect possible genomic rearrangements. (MYBPC3 exon numbering according to NG_007667.1, DNA variants were numbered according to reference sequence LRG_386 (identical to reference sequence NM_000256.3) using HGVS nomenclature (www.HGVS.org)). Variants have been submitted to the Leiden Open Variation Database (http://databases.lovd.nl/shared/genes/MYBPC3 (patient IDs: 00019603–00019606).
Literature review
We searched the literature for clinical and molecular data of patients with neonatal cardiomyopathy and biallelic MYBPC3 variants, focusing on English-language articles published in PubMed and EMBASE between 1995 and January 2013. We identified articles in which the title/abstract included one of the following terms: double MYBPC3 mutations, biallelic MYBPC3 mutations, homozygous or compound heterozygous MYBPC3 mutations or neonatal hypertrophic cardiomyopathy. We then assessed all the references cited by these articles. In our analysis, we only used reports if pathogenic homozygous or compound heterozygous MYBPC3 variants were identified.
Results
Table 1 summarizes the characteristics of the patients and their parents.
Patient 1
Patient 1 was hospitalized at age 5 weeks because of feeding problems and cyanosis. X-thorax showed a grossly enlarged heart. Echocardiography revealed a moderately dilated left ventricle with severe systolic dysfunction. The apical wall of the left ventricle was excessively thickened with prominent hypertrabeculation. The left and right atria showed mild dilatation. A small secundum atrial septal defect (ASD) was also present. Within 2 weeks the X-thorax and echocardiography revealed further enlargement of the heart (heart–thorax ratio±0.7) with severe left ventricular hypertrophy and deep trabeculations in the apex (interventricular septum in diastole (IVSd) 12 mm, Z+10; left ventricular posterior wall in diastole (LVPWd) 10 mm, Z+10) and a fractional shortening (FS) of the left ventricle of 10% (left ventricular internal dimension (LVID) 23/26). The child died from cardiac failure at age 12 weeks. Macroscopic examination of the heart revealed severe cardiomegaly and dilatation with a total weight of 115 g (normal weight at this age, 30 g). Right ventricular thickening was noted, especially of the posterior wall. The anterior wall of the left ventricle was severely thickened and showed abnormal trabeculation and multiple intertrabecular recesses as seen in noncompaction cardiomyopathy. Secundum ASD was confirmed. No other congenital malformations were found. Microscopic examination of cardiac tissue (Figure 1a) showed myofibrillar disarray in both the ventricular septum and the left ventricular wall. Hypertrophic myocytes with a diameter of 20–30 μm (normal 12 μm) and multiple vacuoles on electron microscopy suggested glycogenosis (Figure 1b). As the echocardiographic images and ECG did not suggest Pompe disease and urine oligosaccharide analysis was normal, no α-glucosidase enzyme or molecular assay was performed.

(a) Microscopic postmortem examination of heart muscle from patient 1. Hypertrophic myocytes with myofibrillar disarray typical of HCM due to sarcomeric protein variations were present, albeit without a significant amount of interstitial fibrosis (hematoxylin and eosin staining, magnification × 100). (b) Electron micrograph showing a large, irregular vacuole.
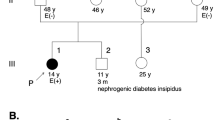
MYBPC3 mutation analysis revealed compound heterozygosity for the pathogenic mutations c.2373dup p.(Trp792fs) and c.2827C>T p.(Arg943*). Both pathogenic mutations are known founder mutations within the Dutch HCM population.10,11 Neither parent had cardiac symptoms. Initial echocardiography revealed no abnormalities in the 32-year-old mother, who was heterozygous for c.2373dup, nor in the 31-year-old father, heterozygous for c.2827C>T. ECG in the father showed mild repolarization abnormalities (ST elevation of 0.5 mm in V1, followed by a negative T in V1–V4). Re-evaluation after 7 years revealed moderate septal hypertrophy (HCM) with an interventricular septum of 14 mm in the mother, and interventricular septal measurements at the upper limit of the normal range (12 mm) in the father. The family history of both parents was positive for HCM and sudden death (Figure 2a).

Pedigrees of patients 1, 3, and 4.
Patient 2
Patient 2 had feeding problems in the first weeks of life and dyspnea and mild hypotonia were noticed at age 4 weeks. A grossly enlarged heart was observed on the X-thorax (heart–thorax ratio 0.8%) and low oxygen saturation blood levels were found. Echocardiography revealed moderately dilated ventricles with severe diastolic and systolic dysfunction. Hypertrabeculation with flow perfused intertrabecular recesses was present in both ventricles, including the apical walls (Figure 3). An apical muscular ventricular septal defect (VSD) was visualized, although deep recessal flow complicated interpretation of the images. Unfortunately, owing to the critically ill status of the patient, there are no reliable M-mode measurements of the left ventricle. A muscle computed tomography scan showed no signs of atrophy, and a muscular biopsy revealed no congenital myopathy. His condition deteriorated at age 12 weeks and he died.

Echocardiographic studies in patient 2 showing cardiomyopathy with LVNC. The four-chamber view shows excessive trabeculation (arrows) at the apical right ventricular wall, whereas the left ventricular wall also showed numerous deep trabeculae and recesses.
In this case too, we identified compound heterozygosity for the pathogenic c.2373dup p.(Trp792fs) and c.2827C>T p.(Arg943*) mutations in the MYBPC3 gene. The parents agreed neither to an autopsy on the child nor to cardiologic evaluation for themselves or pedigree analysis.
Patient 3
The third proband presented with severe feeding problems and signs of cardiac decompensation at age 7 weeks. Cardiomegaly was seen on chest X-ray, and echocardiography showed HCM with dilated ventricles. M-Mode measurements were unavailable. Noncompaction was not a clear feature, but could not be excluded. There was a small foramen ovale and mitral valve insufficiency. The condition of the patient deteriorated owing to a viral infection and she died at age 12 weeks. The parents did not give permission for autopsy and, at that time, molecular analysis of genes involved in cardiomyopathy was not available. Cardiac analyses of first-degree relatives were normal.
Eleven years later, a cousin of the father presented with cardiac arrest due to HCM. He had a heterozygous pathogenic c.2373dup p.(Trp792fs) mutation in MYBPC3. We subsequently confirmed homozygosity for the c.2373dup variant in DNA isolated from stored fibroblasts from the deceased proband. Both parents of the proband were heterozygous for this truncating variant (Figure 2b). Cardiac re-evaluation showed only slightly reduced ECG voltages in the father (age 38) and borderline hypertrophy (septal thickness 11 mm) in the mother (age 41).
Patient 4
Patient 4 presented with dyspnea, hypotension, feeding difficulties, and failure to thrive at age 6 weeks. Echocardiography showed severe HCM with features of noncompaction, and severe diastolic and systolic left ventricular dysfunction (IVSd 11 mm, Z+10; LVPWd 10 mm, Z+10, FS 13% (LVID 17/20) and left ventricular mass 255 g/m2). A secundum ASD with a left–right shunt was also observed. One week later she died after an unsuccessful resuscitation. Homozygosity for the c.2827C>T p.(Arg943*) nonsense mutation in MYBPC3 was found. Both parents were heterozygous for this pathogenic mutation.
Her mother was diagnosed with postpartum HCM with diastolic dysfunction, confirmed by cardiac MRI, at age 26. Echocardiography of the father was normal; ECG showed a right bundle branch block. A paternal uncle of patient 4 had died aged 3 months of unknown cause (Figure 2c).
Literature review
The results of our literature review are summarized in Table 2, showing 21 cases (including our four cases) of double-truncating pathogenic mutations, all presented neonatally or in early childhood. Fifteen died before the age of 1 year; the others had either undergone transplantations or were severely affected. Structural defects were detected in 13 cases, but neither hypertrabecularization nor deep intertrabecular recesses were described in any of the previously reported cases.
Discussion
We describe four unrelated newborns with severe cardiomyopathy due to compound heterozygosity or homozygosity for pathogenic truncating mutations in the MYBPC3 gene. All of them died soon after birth. Although initially their cardiomyopathies were described as severe atypical HCM, HCM with features of left ventricular noncompaction (LVNC) was considered after re-evaluation of serial ultrasounds and pathologic examination in three of them. Like HCM, LVNC is genetically heterogeneous.9,12 Recently, LVNC was shown to be mainly caused by heterozygous variants in genes encoding sarcomeric proteins, including MYH7, ACTC1, and TNNT2.9,12 This study confirms our previous results demonstrating that pathogenic variants in another sarcomeric protein gene, MYBPC3, can also lead to LVNC (OMIM number #615396, LVNC10).9,13 MYBPC3 mutations in HCM lead to an altered primary contractile function. Whether contractile dysfunction is the mechanism that links mutant sarcomere protein to the morphologic features of LVNC is still uncertain.13
In the majority of patients with familial cardiomyopathy owing to a variant in one of the genes encoding sarcomeric proteins, a single autosomal dominant pathogenic mutation is found. In contrast, our four patients were compound heterozygotes (two patients) or homozygous (two patients) for two truncating MYBPC3 variants, suggesting a cumulative effect. Pathogenic mutations in the MYBPC3 gene are one of the most common genetic causes of HCM in many populations, found in 20–40% of individuals with HCM.14,15 Autosomal dominant variants in the MYBPC3 gene, which are mostly truncating pathogenic mutations and sometimes missense variants, give rise to HCM with an age of onset after the third decade, moderate left ventricular hypertrophy, and a favorable prognosis.16 As variants in MYBPC3 (and MYH7) are the most common genetic cause of familial HCM,6 compound heterozygous or homozygous variants should be considered in a neonate who presents with severe HCM or LVNC, even in the absence of symptoms in family members. This is illustrated by patients 1 and 2 who were compound heterozygotes for the two most common Dutch pathogenic founder mutations for HCM: c.2373dup p.(Trp792fs) and c.2827C>T p.(Arg943*).10,11 Patients 3 and 4 were homozygous for the c.2373dup p.(Trp792fs) and c.2827C>T p.(Arg943*) mutations, respectively.
The pathogenic c.2373dup p.(Trp792fs) mutation accounts for approximately one-fifth of all HCM cases in the Netherlands. It is an important founder mutation in the Dutch HCM population and is also present in other populations.10,11,14 It creates a new aberrant splice donor site leading to skipping of exon 24, resulting in a frameshift after p.Gln791 and a premature stop codon.17 No truncated protein product from the c.2373dup p.(Trp792fs) allele could be detected in the sarcomere-using antibodies, suggesting that the truncated protein was unstable, or the aberrant transcript was degraded by cell surveillance mechanisms such as nonsense-mediated decay.18
The c.2827C>T p.(Arg943*) nonsense mutation was found in three of our patients and is located in exon 27, leading to a premature stop codon and protein truncation beyond domain C7 of MYBPC3. It is thought to lead to reduction in MYBPC3 owing to protein instability and/or loss of the C-terminus of MYBPC3 that binds myosin thick filaments and titin, which is required to incorporate MYBPC3 normally into the A-band of the sarcomere.19 Consequently, the MYBPC3 protein was not expected to be incorporated into the sarcomere in any of these four patients. This may explain the early and severe presentation of their cardiomyopathy.
In larger series, ~3–5% of adult HCM patients prove to be compound or double heterozygotes for two disease-causing variants in the same or different sarcomeric protein genes.14,15 However, most of these are missense variants and their pathogenic nature is not always easy to establish. In these adult cases, a more severe HCM phenotype is generally seen, characterized by an earlier age of onset around the second decade or during childhood (Table 2).6,14,15,20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30 In a recent study on sarcomeric protein gene variants in childhood-onset HCM, 6 out of 84 children (7%) had compound variants.6 This suggests that a gene–dosage effect might be responsible for manifestations at a younger age. As Table 2 shows, all patients with two functional null alleles died within the first year of life. Patients with a missense variant and a truncating pathogenic mutation, or patients with two missense variants in MYBPC3, had a milder phenotype and an onset of cardiomyopathy at a more advanced age. This might be explained by some residual activity of the MYBPC3 protein.
All cases with biallelic truncating MYBPC3 mutations present as neonates. Nevertheless, we do expect prenatal onset of HCM in these patients. One of the reasons that prenatal presentation has not yet been reported, could be that prenatal ultrasounds in the Netherlands, as well as in most other European countries, are not performed at a regular basis after 22 weeks of gestation. Also, mutation analysis of sarcomeric genes is not always performed in pregnancies of fetuses with cardiomyopathy, and some of these fetuses might die before a molecular diagnosis can be made. In addition, prenatal circulation is different from postnatal circulation, after the fetus is disconnected from the umbilical vessels. Owing to the loss of tremendous blood flow through the placenta, the systemic vascular resistance at birth doubles. As resistance rises, aortic and left ventricular pressure increase. The neonatal heart adapts (remodels) to sudden increased systolic pressures following birth, by increasing ventricular wall thickness and stiffness (ie tensile strength). This is a result of a twofold increase in the number of fibroblasts and the formation, compaction, and alignment of collagen fibrils that envelop myocytes. We hypothesize that absence of MYBPC3 protein would impair this process, leading to HCM with features of noncompaction in neonates.
Few neonatal cases with severe cardiomyopathy owing to homozygous or compound heterozygous truncating pathogenic mutations in MYBPC3 have been described.14,31, 32, 33, 34, 35 A homozygous truncating pathogenic splice site variant c.3330+2T>G in MYBPC3 was reported in three neonates with severe HCM who all died at an average age of 3–4 months in a consanguineous Old Order Amish pedigree with severe HCM.31 This variant was also reported in another cohort of 10 neonates with severe infantile HCM of Old Order Amish descent, suggesting a founder effect.33 Of note, several of the affected Amish neonates with homozygous MYBPC3 truncating variants also presented with septal defects including apical muscular VSD, ASD, and patent ductus arteriosus. Septal defects were also present in the neonates with severe HCM owing to compound heterozygous truncating pathogenic mutations described by Lekanne et al32 and in three of our cases. Different congenital heart malformations (septal defects, patent ductus arteriosus, aortic aneurysm, and Ebstein anomaly) have been reported in families with pathogenic mutations in sarcomeric protein genes, including MYBPC3, MYH6, MYH7, MYH11, and ACTC1.31,36, 37, 38 In children with noncompaction cardiomyopathy, Tsai et al39 showed that 78% had a congenital heart defect. These data suggest that sarcomeric cardiac muscle proteins are not only involved in cardiomyopathies but also in congenital heart malformations.40 Table 2 shows that only patients with double-truncating pathogenic mutations have a septal defect, suggesting that functional MYBPC3 protein has a crucial role in septal development. Septal defects have not been reported in patients with missense variants, or with a missense variant in combination with a truncating variant. In conclusion, the absence of functional MYBPC3 from the sarcomere can lead to a phenotype of severe HCM with features of ventricular noncompaction and septal defects, which appears to be lethal in the postnatal period.
References
Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE : Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation 1995; 92: 785–789.
Alcalai R, Seidman JG, Seidman CE : Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. J Cardiovasc Electrophys 2008; 19: 104–110.
Bos JM, Ommen SR, Ackerman MJ : Genetics of hypertrophic cardiomyopathy: one, two, or more diseases? Curr Opin Cardiol 2007; 22: 193–199.
Lopes LR, Rahman MS, Elliott PM : A systematic review and meta-analysis of genotype-phenotype associations in patients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations. Heart 2013; 99: 1800–1811.
Daw EW, Chen SN, Czernuszewicz G et al: Genome-wide mapping of modifier chromosomal loci for human hypertrophic cardiomyopathy. Hum Molec Genet 2007; 16: 3463–3471.
Morita H, Rehm HL, Menesses A et al: Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med 2008; 358: 1899–1908.
Grenier MA, Osganian SK, Cox GF et al: Design and implementation of the North American Pediatric Cardiomyopathy Registry. Am Heart J 2000; 139: S86–S95.
Jenni R, Oechslin EN, van der Loo B : Isolated ventricular non-compaction of the myocardium in adults. Heart 2007; 93: 11–15.
Hoedemaekers YM, Caliskan K, Michels M et al: The importance of genetic counseling, DNA diagnostics, and cardiologic family screening in left ventricular noncompaction cardiomyopathy. Circ Cardiovasc Genet 2010; 3: 232–239.
Alders M, Jongbloed R, Deelen W et al: The 2373insG mutation in the MYBPC3 gene is a founder mutation, which accounts for nearly one-fourth of the HCM cases in the Netherlands. Eur Heart J 2003; 24: 1848–1853.
Christiaans I, Nannenberg EA, Dooijes D et al: Founder mutations in hypertrophic cardiomyopathy patients in the Netherlands. Neth Heart J 2010; 18: 248–254.
Klaassen S, Probst S, Oechslin E et al: Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation 2008; 117: 2893–2901.
Probst S, Oechslin E, Schuler P et al: Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circ Cardiovasc Genet 2011; 4: 367–374.
Richard P, Charron P, Carrier L et al: Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003; 107: 2227–2232.
Van Driest SL, Vasile VC, Ommen SR et al: Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol 2004; 44: 1903–1910.
Niimura H, Bachinski LL, Sangwatanaroj S et al: Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med 1998; 338: 1248–1257.
Moolman JA, Reith S, Uhl K et al: A newly created splice donor site in exon 25 of the MyBP-C gene is responsible for inherited hypertrophic cardiomyopathy with incomplete disease penetrance. Circulation 2000; 101: 1396–1402.
van Dijk SJ, Dooijes D, dos Remedios C et al: Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 2009; 119: 1473–1483.
Rottbauer W, Gautel M, Zehelein J et al: Novel splice donor site mutation in the cardiac myosin-binding protein-C gene in familial hypertrophic cardiomyopathy. Characterization Of cardiac transcript and protein. J Clin Invest 1997; 100: 475–482.
Maron BJ, Maron MS, Semsarian C : Double or compound sarcomere mutations in hypertrophic cardiomyopathy: a potential link to sudden death in the absence of conventional risk factors. Heart Rhythm 2012; 9: 57–63.
Nanni L, Pieroni M, Chimenti C et al: Hypertrophic cardiomyopathy: two homozygous cases with "typical" hypertrophic cardiomyopathy and three new mutations in cases with progression to dilated cardiomyopathy. Biochem Biophys Res Comm 2003; 309: 391–398.
Garcia-Castro M, Reguero JR, Alvarez V et al: Hypertrophic cardiomyopathy linked to homozygosity for a new mutation in the myosin-binding protein C gene (A627V) suggests a dosage effect. Int J Cardiol 2005; 102: 501–507.
Ingles J, Doolan A, Chiu C, Seidman J, Seidman C, Semsarian C : Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. J Med Genet 2005; 42: e59.
Otsuka H, Arimura T, Abe T et al: Prevalence and distribution of sarcomeric gene mutations in Japanese patients with familial hypertrophic cardiomyopathy. Circ J 2012; 76: 453–461.
Dellefave LM, Pytel P, Mewborn S et al: Sarcomere mutations in cardiomyopathy with left ventricular hypertrabeculation. Circ Cardiovasc Genet 2009; 2: 442–449.
Girolami F, Ho CY, Semsarian C et al: Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J Am Coll Cardiol 2010; 55: 1444–1453.
Marziliano N, Merlini PA, Vignati G et al: A case of compound mutations in the MYBPC3 gene associated with biventricular hypertrophy and neonatal death. Neonatology 2012; 102: 254–258.
Garcia-Castro M, Coto E, Reguero JR et al: [Mutations in sarcomeric genes MYH7, MYBPC3, TNNT2, TNNI3, and TPM1 in patients with hypertrophic cardiomyopathy]. Rev Esp Cardiol 2009; 62: 48–56.
Saltzman AJ, Mancini-DiNardo D, Li C et al: Short communication: the cardiac myosin binding protein C Arg502Trp mutation: a common cause of hypertrophic cardiomyopathy. Circ Res 2010; 106: 1549–1552.
Bashyam MD, Purushotham G, Chaudhary AK et al: A low prevalence of MYH7/MYBPC3 mutations among familial hypertrophic cardiomyopathy patients in India. Molec Cell Biochem 2012; 360: 373–382.
Xin B, Puffenberger E, Tumbush J, Bockoven JR, Wang H : Homozygosity for a novel splice site mutation in the cardiac myosin-binding protein C gene causes severe neonatal hypertrophic cardiomyopathy. Am J Med Genet A 2007; 143A: 2662–2667.
Lekanne Deprez RH, Muurling-Vlietman JJ, Hruda J et al: Two cases of severe neonatal hypertrophic cardiomyopathy caused by compound heterozygous mutations in the MYBPC3 gene. J Med Genet 2006; 43: 829–832.
Zahka K, Kalidas K, Simpson MA et al: Homozygous mutation of MYBPC3 associated with severe infantile hypertrophic cardiomyopathy at high frequency amongst the Amish. Heart 2008; 94: 1326–1330.
Ortiz MF, Rodriguez-Garcia MI, Hermida-Prieto M et al: A homozygous MYBPC3 gene mutation associated with a severe phenotype and a high risk of sudden death in a family with hypertrophic cardiomyopathy. Rev Esp Cardiol 2009; 62: 572–575.
Tajsharghi H, Leren TP, Abdul-Hussein S et al: Unexpected myopathy associated with a mutation in MYBPC3 and misplacement of the cardiac myosin binding protein C. J Med Genet 2010; 47: 575–577.
Ching YH, Ghosh TK, Cross SJ et al: Mutation in myosin heavy chain 6 causes atrial septal defect. Nat Genet 2005; 37: 423–428.
Zhu L, Vranckx R, Khau Van Kien P et al: Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet 2006; 38: 343–349.
Matsson H, Eason J, Bookwalter CS et al: Alpha-cardiac actin mutations produce atrial septal defects. Hum Molec Genet 2008; 17: 256–265.
Tsai SF, Ebenroth ES, Hurwitz RA, Cordes TM, Schamberger MS, Batra AS : Is left ventricular noncompaction in children truly an isolated lesion? Pediatr Cardiol 2009; 30: 597–602.
Wessels MW, Willems PJ : Mutations in sarcomeric protein genes not only lead to cardiomyopathy but also to congenital cardiovascular malformations. Clin Genet 2008; 74: 16–19.
Acknowledgements
We thank the families and the referring physicians. We thank Dr KP Dingemans for providing an electronic micrograph, T de Vries Lentsch for graphical support, Dr F van den Heuvel for re-evaluating an echocardiography, and Dr JP van Tintelen and J Senior for editing the text.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Wessels, M., Herkert, J., Frohn-Mulder, I. et al. Compound heterozygous or homozygous truncating MYBPC3 mutations cause lethal cardiomyopathy with features of noncompaction and septal defects. Eur J Hum Genet 23, 922–928 (2015). https://doi.org/10.1038/ejhg.2014.211
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.211
This article is cited by
-
Identification of an elusive spliceogenic MYBPC3 variant in an otherwise genotype-negative hypertrophic cardiomyopathy pedigree
Scientific Reports (2022)
-
Overlap phenotypes of the left ventricular noncompaction and hypertrophic cardiomyopathy with complex arrhythmias and heart failure induced by the novel truncated DSC2 mutation
Orphanet Journal of Rare Diseases (2021)
-
Mutations in genes associated with either myopathy or noncompaction
Herz (2019)
-
Gene therapy strategies in the treatment of hypertrophic cardiomyopathy
Pflügers Archiv - European Journal of Physiology (2019)
-
Untying the knot: protein quality control in inherited cardiomyopathies
Pflügers Archiv - European Journal of Physiology (2019)
