
Abstract
Klinefelter syndrome (47, XXY) is the most frequent genetic cause of male infertility and individuals share the endocrine hallmark of hypergonadotropic hypogonadism. Single-nucleotide polymorphisms located within the FSHB/FSHR gene were recently shown to impact serum follicle-stimulating hormone (FSH) levels and other reproductive parameters in men. The objective of this study was to analyse the effect of FSHB-211G>T (c.−280G>T, rs10835638) as well as FSHR c.2039G>A (rs6166) and FSHR c.−29G>A (rs1394205) on endocrine and reproductive parameters in untreated and testosterone-treated Klinefelter patients. Patients were retrospectively selected from the clientele attending a university-based andrology centre. A total of 309 non-mosaic Klinefelter individuals between 18 and 65 years were included and genotyped for the variants by TaqMan assays. The untreated group comprised 248 men, in which the FSHB −211G>T allele was significantly associated with the reduced serum follicle-stimulating hormone levels (−6.5 U/l per T allele, P=1.3 × 10−3). Testosterone treatment (n=150) abolished the observed association. When analysing patients before and under testosterone treatment (n=89), gonadotropin levels were similarly suppressed independently of the FSHB genotype. The FSHR polymorphisms did not exhibit any significant influence in any group, neither on the endocrine nor reproductive parameters. In conclusion, a hypergonadotropic setting such as Klinefelter syndrome does not mask the FSHB −211G>T genotype effects on the follicle-stimulating hormone serum levels. The impact was indeed more pronounced compared with normal or infertile men, whereas gonadotropin suppression under testosterone treatment seems to be independent of the genotype. Thus, the FSHB −211G>T genotype is a key determinant in the regulation of gonadotropins in different reproductive-endocrine pathopyhsiologies.
Similar content being viewed by others

Introduction
Klinefelter’s syndrome (KS) is the most frequent genetic cause of male infertility.1 The syndrome is caused by a numerical sex chromosomal aberration (47, XXY or higher-grade aneuploidies and mosaicisms). Patients typically display small firm testes and azoospermia,2 while other symptoms including eunuchoid tall stature, gynaecomastia, display a broad interindividual phenotypic variability.3 Due to deficient testicular function and hence the absence of negative feedback regulation by the gonadal steroids and Inhibin B, adult Klinefelter individuals share the endocrine hallmark of hypergonadotropic FSH and luteinzing hormone (LH) levels, often in the presence of low testosterone levels.4, 5, 6 Recently, a single-nucleotide polymorphism (SNP; FSHB c.−280G>T, rs10835638, commonly denoted as FSHB −211G>T, see Materials and methods) in the promoter of the FSHB gene, coding for the β-subunit of FSH, has been observed to be associated with reduced serum FSH levels in men.7, 8, 9, 10 Furthermore, associations with testosterone and other downstream reproductive parameters, that is, lower bi-testicular volume and lower sperm concentration/count have been reported.10 In addition, two SNPs in the FSH receptor, FSHR c.2039G>A (rs6166) and FSHR c.−29G>A (rs1394205), have been shown to impact serum FSH levels in men.11 As Klinefelter individuals display supraphysiological, but highly variable gonadotropin levels, we asked whether an impact of the FSHB −211G>T SNP on gonadotropins exists and if testosterone treatment influences gonadotropin suppression depending on the SNP genotype. To address this, we investigated large cohorts of KS patients who were either treatment naive or under testosterone treatment. Further, we included FSHR c.2039G>A and FSHR c.−29G>A in our analysis and additionally measured Inhibin B and anti-Müllerian hormone (AMH) levels in a subgroup of 52 untreated KS individuals and a control group comprising 50 healthy men to asses Sertoli cell function.
Materials and methods
Study population
All patients were retrospectively selected from the clientele attending the Department of Clinical Andrology, Centre of Reproductive Medicine and Andrology, University Clinic Münster, using the Androbase database.12 Klinefelter individuals between 18 and 65 years were included while excluding patients with higher-grade aneuploidies or mosaicisms. A total of 309 KS individuals were included forming the following groups: the first group comprised of 248 patients who were either treatment naïve or did not receive any testosterone treatment in the previous 12 months (untreated group). A second group comprised of 150 patients who underwent testosterone treatment at the time of hormone measurement (treated group). Around half of these patients were treated with testosterone gel and the other half with quarterly injections of testosterone undecanoate. Both groups shared 89 KS individuals with both a treated and an untreated status. To establish previously unavailable normal ranges for Inhibin B and AMH, 50 male controls (mean age: 42.4 years±14.2, range: 21–68 years) were recruited by advertisement in newspapers, consented specifically and analysed. Health status of controls was not specifically asked for to minimize bias.
Ethics statement
All participants gave informed written consent for evaluation of their clinical data and genetic analysis of their donated DNA samples according to protocols approved by the Ethics Committee of the Medical Faculty in Münster (Az4INie).
Hormone analyses
A venous blood sample was drawn from the cubital vein. FSH and LH were measured by immunofluorometric assays (Autodelfia; Perkin-Elmer, Freiburg, Germany). Serum testosterone (DRG-AURICA-ELISA testosterone kit; DRG-Instruments, Marburg, Germany), AMH and Inhibin B (AMH/Inhibin B Gen II Kit, Beckman Coulter, Sinsheim, Germany) were measured by a commercial ELISA. Intra- and interassay coefficients of variation were less than 5% and less than 10%, respectively. Free testosterone was calculated from total testosterone and sex hormone-binding globulin (SHBG) concentrations.13
Genotyping
Genomic DNA was extracted from EDTA-preserved blood using the FlexiGene DNA extraction kit (QIAGEN, Düsseldorf, Germany). Genotyping was performed with the StepOnePlus Real-Time PCR-System, the TaqMan GTXpress Master Mix and a TaqMan genotyping assay mix customized for the FSHB c.−280G>T (rs10835638, NC_000011.10:g.30230805G>T and NM_000510.2:c.−280G>T, most commonly denoted as FSHB −211G>T referring to the position upstream of the mRNA transcription start site, which is predicted 69 bp upstream of the ATG codon), FSHR −29G>A (rs1394205, NC_000002.12:g.49154446C>T and NM_000145.3:c.−29G>A) and FSHR c.2039G>A (rs6166, NC_000002.12:g.48962782C>T and NM_000145.3:c.2039G>A; because the common allele in most populations is A, this SNP has frequently been denoted as c.2039A>G) SNPs (Applied Biosystems, Carlsbad, CA, USA). The thermal cycling conditions were as follows: 2 min at 50 °C followed by 15 min at 95 °C and 35 cycles of 15 s at 95 °C plus 1 min at 60 °C. All genotyping results have been submitted to dbSNP database (ss ID: ss1056609682, ss1052336034 and ss1052336035, respectively; URL: http://www.ncbi.nlm.nih.gov/SNP/snp_ss.cgi?subsnp_id=1056609682, http://www.ncbi.nlm.nih.gov/SNP/snp_ss.cgi?subsnp_id=1052336034 and http://www.ncbi.nlm.nih.gov/SNP/snp_ss.cgi?subsnp_id=1052336035, respectively). rs10835638 was additionally reported to ClinVar database (Accession SCV000165958; URL: http://www.ncbi.nlm.nih.gov/clinvar/?term=SCV000165958).
Statistics
All analyses were performed using Stata/SE (StataCorp LP, Version 11.2; College Station, TX, USA) using additional Stata subroutines.14 The HWSNP subroutine was used to test for deviation from the Hardy–Weinberg equilibrium and the GENCC subroutine to test for differences in allele frequencies. QTLSNP was used for linear regression to test the hypothesis of equality of the means of parameters across genotypes, including continuous covariate adjustment. Although the additive model assumes a cumulative allele effect in a dose-dependent manner, in the recessive model strong effects are only expected among minor allele homozygotes. As values, except estradiol, did not follow a Gaussian distribution, data were log-transformed to approximate a normal distribution. P<0.05 was considered statistically significant.
Results
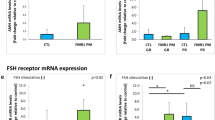
All SNP genotypes were found to be in Hardy–Weinberg equilibrium for the study group (P=NS). Age and BMI differed significantly between the FSHB −211G>T genotype groups (Tables 1 and 2) and were introduced as covariates into the model. Regarding the untreated group, the FSHB −211 T allele was significantly associated with reduced serum FSH levels (additive model: −6.5 U/l per T allele, P=1.3 × 10−3, recessive model: TT homozygosity-effect: −10.1 U/l, P=6.6 × 10−3). No other endocrine or reproductive parameter, for example, bi-testicular volume, was significantly associated with the FSHB −211G>T genotype (Table 1 and Figure 1a). Testosterone treatment of KS patients abolished the previously observed association, although a clear gradient over the three genotypes was observed (Table 2 and Figure 1b). Investigation of each testosterone-agent subgroup alone did not show any significant influences of the SNP (data not shown). LH displayed a non-significant and less pronounced gradient over the three genotypes in the untreated group, which was no longer detectable in the treated group (Tables 1 and 2). When comparing the untreated and treated status in the subgroup of 89 men, mean FSH suppression by testosterone treatment accounted for 49.7%±38.8 among GG-homozygotes, 44.0%±39.8 among GT heterozygotes and 46.0%±31.9 among TT homozygotes and was comparable between genotype groups (P=NS). FSHR c.2039G>A and FSHR c.−29G>A did not exhibit any further significant influence on any parameter neither in the untreated (Supplementary Tables 1 and 2) nor in the treated group (data not shown). Notably, a trend for lower FSH in the untreated FSHR c.−29 A-Allele group was observed. Comparable to a previous study,15 we found AMH and Inhibin B levels in KS individuals strongly reduced (mean: 0.44 ng/ml and 1.5 pg/ml, respectively) in comparison with controls (mean: 5.43 ng/ml and 195 pg/ml, respectively). Only half of the KS patients had AMH levels above the assay detection limit (Supplementary Figure 1) and only two patents displayed measurable Inhibin B levels. No association with the SNPs’ genotypes was found.

Box and whisker plots of FSH serum concentrations according to FSHB −211G>T genotype of the untreated (a) and the treated group (b). Boxes contain 50% of the distribution, whiskers extend to 5–95%; lines in boxes represent the median.
Discussion
The present study evaluated a possible impact of FSHB −211G>T, FSHR c.2039G>A and FSHR c.−29G>A SNPs on the endocrine phenotype in Klinefelter individuals and verified an association of FSHB −211G>T with reduced FSH levels even in a hypergonadotropic setting consistently with previous observations in men.7, 8, 9, 10 A relation of testicular size with FSHB/FSHR genotypes could not be observed, most likely due to the predominant azoospermia in KS patients.
Since the first clinical study, showing an impact of the FSHB -211G>T SNP on FSH levels and other reproductive parameters in a male cohort,7 this SNP has become subject to extensive research in both men and women.7, 8, 9, 10, 16, 17 Recent studies also addressed the clinical use of this genetic marker in male infertility treatment.9, 10 Concerning the function of this SNP, a recent in vitro study identified an altered LHX3 binding site in the human FSHB promoter, encompassing the −211G>T SNP, to be potentially responsible for the reduced transcriptional activity of the FSHB T-allele variant.18 Although LHX3 has been shown to be an essential factor in the regulation of gonadotropin subunit expression,19 it is still unclear how the transcription factor exerts its effect on FSHB transcription. Assuming that the net production of FSHB is limited in the TT genotype, it obviously does not preclude the synthesis of very high levels of FSH as observed in KS patients. Thus, a scenario in which the basal synthesis is hampered, but stimulation at a reduced maximum net amount of FSHB is possible, seems to be very likely. The TT genotype does not seem to impair the negative feedback regulation of the FSHB gene, as testosterone treatment not only suppresses FSH levels, but moreover also abolishes difference between all FSHB genotypes. Thus, the observed effects of testosterone treatment are mainly mediated via a presumably changed hypothalamic GnRH pulse frequency/amplitude effect, which in turn reduces the synthesis rate of both gonadotropins. The strongly reduced or undetectable Inhibin B and AMH levels in KS seem to reflect either an impaired Sertoli cell function (low AMH) and as consequence thereof lacking spermatogenesis (low Inhibin B levels) or might indicate a substantial defect in hormone accessibility of FSH to Sertoli cells. This may be due to a changed testicular architecture as recently shown in KS patients and a mouse model.20 The investigated FSH receptor SNPs had no significant impact on FSH levels probably due to the lack of feedback mechanism.
We found the effect of the T-allele variant of the −211G>T SNP in this cohort of untreated hypergonadotropic individuals more visible than in previous studies on male cohorts, resulting in a decrease of FSH of about 60% (GG vs TT homozygotes). Unfortunately, the groups – primarily the TT group – differ significantly in age and BMI. As these two variables can be independent determinants of gonadotropins and reproductive parameters,21, 22 we used them for covariate adjustment to control for confounding. Collectively, investigating the hypergonadotropic setting in KS, our results underline the role of this SNP in altering FSHB expression.
We further showed that the effect of the SNP on FSH levels is no longer significant in the testosterone-treated group. Around half of these patients were treated with testosterone gel and the other half with quarterly injections of testosterone undecanoate, resulting in broad interindividual variance of gonadotropin suppression. Investigation of each group alone did not exhibit any significance influences of the SNP, probably due to the reduced group sizes. The reason for the loss of significance rather seems to be the broad interindividual variance of gonadotropin suppression by testosterone than the loss of the impact of the SNP. In addition, testosterone substitution was sufficient to suppress elevated gonadotropin levels comparably across all genotypes.
Our study raises important questions: first, it would be interesting to study whether KS patients carrying different FSHB genotypes show differences in testicular phenotype. In the virtual absence of spermatogenesis, the direct effect of FSH on Sertoli cell number during (pre-)pubertal development could be investigated and it would also be interesting to see whether patients with the FSHB TT genotype have less Sertoli cells. Second, it would be interesting whether the observed effect is also detectable in elderly women, another setting with hypergonadotropism, where it might modulate the onset of menopause.
References
Tüttelmann F, Gromoll J : Novel genetic aspects of Klinefelter‘s syndrome. Mol Hum Reprod 2010; 16: 386–395.
Kamischke A, Baumgardt A, Horst J, Nieschlag E : Clinical and diagnostic features of patients with suspected Klinefelter syndrome. J Androl 2003; 24: 41–48.
Bojesen A, Juul S, Gravholt CH : Prenatal and postnatal prevalence of Klinefelter syndrome: a national registry study. J Clin Endocrinol Metab 2003; 88: 622–626.
Means AR, Fakunding JL, Huckins C, Tindall DJ, Vitale R : Follicle-stimulating hormone, the Sertoli cell, and spermatogenesis. Recent Prog Horm Res 1976; 32: 477–527.
Peterson NT Jr, Midgley AR Jr, Jaffe RB : Regulation of human gonadotropins. 3. Luteinizing hormone and follicle stimulating hormone in sera from adult males. J Clin Endocrinol Metab 1968; 28: 1473–1478.
Lanfranco F, Kamischke A, Zitzmann M, Nieschlag E : Klinefelter’s syndrome. Lancet 2004; 364: 273–283.
Grigorova M, Punab M, Ausmees K, Laan M : FSHB promoter polymorphism within evolutionary conserved element is associated with serum FSH level in men. Hum Reprod 2008; 23: 2160–2166.
Grigorova M, Punab M, Poolamets O et al: Increased prevalance of the -211 T allele of follicle stimulating hormone (FSH) beta subunit promoter polymorphism and lower serum FSH in infertile men. J Clin Endocrinol Metab 2010; 95: 100–108.
Ferlin A, Vinanzi C, Selice R, Garolla A, Frigo AC, Foresta C : Toward a pharmacogenetic approach to male infertility: polymorphism of follicle-stimulating hormone beta-subunit promoter. Fertil Steril 2011; 96: e1342.
Tüttelmann F, Laan M, Grigorova M, Punab M, Sober S, Gromoll J : Combined Effects of the Variants FSHB -211G/T and FSHR 2039A>G on Male Reproductive Parameters. J Clin Endocrinol Metab 2012; 97: 3639–3647.
Grigorova M, Punab M, Punab AM et al: Reproductive physiology in young men is cumulatively affected by FSH-action modulating genetic variants: FSHR -29G/A and c.2039 A/G, FSHB -211G/T. PLoS One 2014; 9: e94244.
Tüttelmann F, Luetjens CM, Nieschlag E : Optimising workflow in andrology: a new electronic patient record and database. Asian J Androl 2006; 8: 235–241.
Vermeulen A, Verdonck L, Kaufman JM : A critical evaluation of simple methods for the estimation of free testosterone in serum. J Clin Endocrinol Metab 1999; 84: 3666–3672.
Cleves MA : Exploratory analysis of single nucleotide polymorphism (SNP) for quantitative traits. Stata J 2005; 5: 141–153.
Aksglaede L, Christiansen P, Sorensen K et al: Serum concentrations of anti-Mullerian hormone (AMH) in 95 patients with Klinefelter syndrome with or without cryptorchidism. Acta Paediatr 2011; 100: 839–845.
Schüring AN, Busch AS, Bogdanova N, Gromoll J, Tüttelmann F : Effects of the FSH-beta-subunit promoter polymorphism -211G->T on the hypothalamic-pituitary-ovarian axis in normally cycling women indicate a gender-specific regulation of gonadotropin secretion. J Clin Endocrinol Metab 2013; 98: E82–E86.
La Marca A, Papaleo E, Alviggi C et al: The combination of genetic variants of the FSHB and FSHR genes affects serum FSH in women of reproductive age. Hum Reprod 2013; 28: 1369–1374.
Benson CA, Kurz TL, Thackray VG : A human FSHB promoter SNP associated with low FSH levels in men impairs LHX3 binding and basal FSHB transcription. Endocrinology 2013; 154: 3016–3021.
Pfaeffle RW, Savage JJ, Hunter CS et al: Four novel mutations of the LHX3 gene cause combined pituitary hormone deficiencies with or without limited neck rotation. J Clin Endocrinol Metab 2007; 92: 1909–1919.
Tüttelmann F, Damm OS, Luetjens CM et al: Intratesticular testosterone is increased in men with Klinefelter syndrome and may not be released into the bloodstream owing to altered testicular vascularization - a preliminary report. Andrology 2014.
Chavarro JE, Toth TL, Wright DL, Meeker JD, Hauser R : Body mass index in relation to semen quality, sperm DNA integrity, and serum reproductive hormone levels among men attending an infertility clinic. Fertil Steril 2010; 93: 2222–2231.
Moroz EV, Verkhratsky NS : Hypophyseal-gonadal system during male aging. Arch Gerontol Geriatr 1985; 4: 13–19.
Acknowledgements
We would like to thank Nicole Terwort, Sabine Forsthoff and Reinhild Sandhowe for technical assistance. The study was in part supported by a grant from the IZKF of the University of Münster (CRA03/09).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Busch, A., Tüttelmann, F., Zitzmann, M. et al. The FSHB −211G>T variant attenuates serum FSH levels in the supraphysiological gonadotropin setting of Klinefelter syndrome. Eur J Hum Genet 23, 700–703 (2015). https://doi.org/10.1038/ejhg.2014.142
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.142

{kind=link}