
Abstract
Classical Beckwith–Wiedemann syndrome (BWS) was diagnosed in two sisters and their male cousin. The children’s mothers and a third sister were tall statured (178, 185 and 187 cm) and one had mild BWS features as a child. Their parents had average heights of 173 cm (mother) and 180 cm (father). This second generation tall stature and third generation BWS correlated with increased methylation of the maternal H19/IGF2-locus. The results were obtained by bisulphite treatment and subclone Sanger sequencing or next generation sequencing to quantitate the degree of CpG-methylation on three locations: the H19 promoter region and two CTCF binding sites in the H19 imprinting control region (ICR1), specifically in ICR1 repeats B1 and B7. Upon ICR1 copy number analysis and sequencing, the same maternal point variant NCBI36:11:g.1979595T>C that had been described previously as a cause of BWS in three brothers, was found. As expected, this point variant was on the paternal allele in the non-affected grandmother. This nucleotide variant has been shown to affect OCTamer-binding transcription factor-4 (OCT4) binding, which may be necessary for maintaining the unmethylated state of the maternal allele. Our data extend these findings by showing that the OCT4 binding site mutation caused incomplete switching from paternal to maternal ICR1 methylation imprint, and that upon further maternal transmission, methylation of the incompletely demethylated variant ICR1 allele was further increased. This suggests that maternal and paternal ICR1 alleles are treated differentially in the female germline, and only the paternal allele appears to be capable of demethylation.
Similar content being viewed by others


Introduction
Beckwith–Wiedemann syndrome (BWS, OMIM no. 130650) is an overgrowth condition caused by epigenetic or genetic alterations in the imprinted H19/IGF2-KCNQ1/CDKN1C locus, spanning nearly 1 Mb in 11p15.5.1 The two major causes of BWS are increased IGF2 expression or decreased CDKN1C expression. The growth inhibitor CDKN1C is normally expressed from the maternal chromosome 11. On the paternal chromosome, inhibition of CDKN1C is associated with the expression of a long noncoding RNA called KCNQ1OT1 or LIT1 antisense to KCNQ1, the potassium channel gene involved in long-QT-syndrome type 1 and Jervell/Lange-Nielsen syndrome. In contrast, the growth factor IGF2 gene is expressed from the paternal chromosome only. On the maternal chromosome, the noncoding RNA gene H19 is expressed instead. The choices between H19 or IGF2 expression, and LIT1 or CDKN1C expression, are regulated epigenetically. IGF2 expression is associated with methylation of the insulator (CTCF) binding sites between H19 and IGF2, also called imprinting centre region 1 (ICR1), and CDKN1C expression is associated with methylation of the LIT1 promoter, also called imprinting centre region 2 (ICR2).
The regulation of 11p15.5 imprinting that causes monoallelic paternal IGF2 expression and monoallelic maternal CDKN1C expression is complex, which also explains why there are many different molecular causes of BWS.2 The most common cause is reduced expression of CDKN1C, which is usually due to sporadically occurring reduction in maternal ICR2 methylation (∼50% of BWS cases), but sometimes associated with maternal CDKN1C mutations (5–10%). The second most common cause is IGF2 overexpression, usually due to paternal uniparental disomy of 11p (∼20% of BWS cases), but also to inappropriate ICR1 methylation on the maternal allele, which inhibits H19 and stimulates IGF2 expression (∼5%).1 In the latter situation, small deletions or mutations in the ICR1 that are likely to disrupt the insulator function of the region have been observed with a high sibling recurrence risk.3, 4, 5, 6, 7, 8 Recently, it was found that such small and overlapping deletions of ICR1 had variable effects on methylation of the maternal ICR1, indicating that maintenance of maternal hypomethylation was partly dependent on the spatial arrangement of the CTCF binding sites.8
Here, we describe a family with a previously reported ICR1 single nucleotide variant (NCBI36:11:g.1979595T>C) in an OCTamer-binding transcription factor-4 (OCT4) binding site and a gradual increase in ICR1 methylation over the next two generations, the first generation being tall statured, the second generation having full-BWS phenotype with Wilms tumours. This family indicates that ICR1 mutations may affect the ability to establish a maternal ICR1 methylation pattern of the paternal allele in female gonads, that is, to demethylate the paternal ICR1 region. Our data also indicate that the maternal and paternal ICR1 alleles are treated differently in the maternal gonads, that is, that the maternal alleles are not demethylated (if methylated) in the maternal germ line, and may later be subject to a passive (stochastic) increase in methylation. To the best of our knowledge, this is the first description of anticipation in an epigenetic syndrome.
Patients and Methods
Family
BWS was diagnosed in two sisters (III-1 and III-2) and their male cousin (III-3, see Figure 1). Elective caesarean section was performed in both sisters due to large babies. Both sisters had classical BWS features including Wilms tumour and visceromegaly. III-1 was born in week 38 with macroglossia and large kidneys, birth weight was 4860 g (860 g>97.5th centile), length 53 cm (97.5th centile). As an infant, she was successfully treated for Wilms tumour with chemotherapy. At age 6 years, an operative tongue reduction was performed. III-2 was born at term with macroglossia and large kidneys, birth weight 5280 g (880 g>97.5th centile). At age 9 months, she was nephrectomised due to Wilms tumour in her right kidney and mild nephroblastomatosis in her left kidney was also detected. The sisters are now 10 and 13 years, and both have good school performances and growth parameters in upper percentiles (III-1 97.5th centile and III-2 95th centile). Their male cousin (III-3, DZ twin) died from medical complications after a caesarean section in week 29. There was marked polyhydramnios. Birth weight was 2130 g (330 g>97.5th centile), length 44 cm, head circumference 28 cm and he had visceromegaly (especially of the kidneys), macroglossia and general subcutaneous oedema. In comparison, his healthy unaffected DZ sister III-4 was 1170 g (5th centile), 38 cm and had the same head circumference at birth. None of the three affected children had neonatal hypoglycaemia, were markedly asymmetric or had transverse creases on their ear helices. The children’s two mothers and the mothers’ sister were tall statured (178, 185 and 187 cm, that is, lengths from the 96th centile and above) with large hands, and at least one had mild BWS features (large tongue, protruding stomach) as a child (II-1 in Figure 1). The sisters’ parents had heights of 173 cm (mother, 85th centile) and 180 cm (father, 75th centile), and none of them had any BWS-like features as infants. II-1 was born at term with a large tongue and birth weight 5 kg (0.4 kg>97.5th centile). II-3 and II-4 are DZ twins born in week 31 with birth weights 1800 g (50th centile) and 2350 g (99th centile), respectively, and II-4 also had a large tongue. All sisters have very mild and asymptomatic scoliosis, no hemihyperplasia and normally sized tongues as adults.

Family pedigree. The degree of methylation of the locus between H19 and ICR1 was investigated by routine MLPA testing and bisulphite subclone sequencing, indicated by the (first ratio/) and (/middle ratio/), respectively. The methylation of the CTCF binding site 6 (CTS6) within the B1 repeat was investigated by bisulphite next generation sequencing, indicated by the (/final ratio) below the pedigree symbols.
Methylation specific multiplex ligation-dependent probe amplification (MS-MLPA) analysis and bisulphite treatment followed by subclone sequencing
Blood DNA samples were obtained from all individuals except III-1 and III-2, where only saliva DNA samples could be obtained. MS-MLPA copy number and methylation analysis of the BWS/Silver-Russell syndrome region on chromosome 11 was done in the routine diagnostic laboratory using the SALSA MLPA ME030 kit version C2 (MRC-Holland, Amsterdam) and following the manufacturer’s instructions. We also obtained saliva DNA samples from II-1 and II-4 to compare with the results of MS-MLPA analysis of blood DNA, and similar levels of methylation was found: 0.7 in saliva DNA from both individuals, compared with 0.71 and 0.75 in their blood DNA (Figure 1). The bisulphite conversion of DNA was performed with Applied Biosystems methylSEQr Bisulphite Conversion Kit (Life Technologies, Carlsbad, CA, USA), according to the manufacturer’s protocol. PCR was performed using primers designed by Methyl Primer Express Software v1.0 (Applied Biosystems, Life Technologies) for bisulphite sequencing-specific PCR. Forward primer: 5′-ATTATTTTGGTTTTTGGTGAGG-3′ (unconverted: 5′-ACCACCTTGGCCTTTGGTGAGG-3′); reverse primer: 5′-ATACCATAAAAATTCCCCCATA-3′ (unconverted: 5′-ATGCCATGGAAATTCCCCCATG-3′) (HG19: chr11:2019867-2020153). M13 tails were added to all the primers as part of our routine to obtain uniform PCR conditions. PCR was performed in a 25 μl reaction containing 1x AmpliTaq Gold 360 Master Mix Forward (Invitrogen, Life Technologies, Carlsbad, CA, USA), 16% 360 CG enhancer (Invitrogen, Life Technologies), 10 μM forward primer and 10 μM reverse primer. The PCR conditions were as follows: denaturation at 95 °C for 5 min, 5 cycles of 95 °C for 30 s, 50 °C for 2 min and 72 °C for 3 min, followed by 35 cycles of 95 °C for 30 s, 58 °C for 1 min and 72 °C for 3 min, and finally a hold at 60 °C for 60 min. The PCR products were then cloned using TOPO TA Cloning Kit for Sequencing (Invitrogen, Life Technologies), following the manufacturer’s instructions. Ten clones were purified using QIAprep Spin Miniprep kit (Qiagen, Hilden, Germany) and sequenced by Sanger sequencing using the BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Life Technologies, Calrsbad, CA, USA) with T3 or T7 primers. Clean up was performed using the using Big DyeX Terminator purification (Applied Biosystems, Life Technologies). The sequences were resolved on a 3730 Genetic Analyser (Applied Biosystems, Life Technologies) and analysed with QUMA (Riken, Japan). To calculate the average methylation level in controls (n=5, including the grandmother of the family) and affected (n=3 per generation, see Figure 1), the average of all measurements per sample was first calculated, and thereafter the mean per group (Table 1). For MS-MLPA, four repeated measurements per sample were done, and for bisulphite sequencing, seven informative positions with differential methylation were measured once per patient (see Supplementary Table 1 and Supplementary Figure 1 with legend for details). Five data points were noninformative because they were highly methylated in all samples with no difference between patients and controls (Supplementary Table 1), and these data points were therefore excluded. The individual MS-MLPA and bisulphite data can also be seen in Figure 1, and mean data with 95% confidence intervals can be found in Table 1.
Bisulphite treatment followed by next generation sequencing
Bisulphite treatment was conducted using the EZ DNA Methylation-Gold Kit (Zymo Research Europe, Freiberg, Germany) according to the manufacturer’s manual. For each individual bisulphite amplicon libraries were generated and sample-specific barcode sequences were added. The amplicons were purified, diluted and clonally amplified in an emulsion PCR before sequencing on the Roche/454 GS junior system was carried out. For subsequent data analysis the Geneious software (Biomatters, Auckland, New Zealand) and BiqAnalyzer HT were used.9 A detailed description has been published elsewhere.8 A minimum of 1058 reads for each sample was obtained. The average conversion rate was 99.0% for CTS1 and 99.1% for CTS6.
ICR1 sequencing
After confirmation of complete 11p15 grandmaternal haplotype segregation with large growth/BWS (Supplementary Figure 2), ICR1 and the H19 proximal promoter (HG19:chr11:2,020,402-2,024,682) were sequenced by standard methods from a product of 4281 bp generated using primers 5′-TGCACATACTTTGCACATGG/CGCTGTGGCTGATGTGTAG-3′, as described;3 further details are available on request. To determine the parental origin of the sequence variant, 200 ng genomic DNA was cleaved using restriction enzyme McrBc (New England Biolabs, Ipswich, MA, USA) following the manufacturer's instructions; then DNA was desalted and concentrated using Amicon 30K microconcentrator columns (Millipore, Billerica, MA, USA) before amplification using primers 5′-CAACACAAGGATCCTAGACC/TCTTCGTATCGGGCCATATC-3′ and Sanger sequencing.
Results
Our family shows dominant inheritance of BWS with a clinical picture that is compatible with anticipation (Figure 1). This impression was in line with the results of the routine MS-MLPA methylation testing of the BWS locus, which showed an increase in ICR1 methylation from normal level (0.49) in the grandmother to ∼0.71 in the second generation with tall stature and ∼0.84 in the third generation (Table 1, Supplementary Table 1), in the three children with classical BWS (Figure 1). These data were reproduced by bisulphite treatment and subclone sequencing to measure the degree of CpG-methylation of the H19 differentially methylated promotor region (Figure 2).

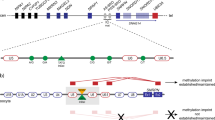
Illustration of the H19/IGF2 ICR1. Above the line approximate positions of CTCF binding sites are marked with green dots, and OCT4 sites are marked with blue dots. The position of a CpG island in the H19 promoter is shown above the line. Below the line the differentially methylated CpG sites investigated by the routine MLPA kit are marked with red dots, and these sites are not included in the segment from the CpG island (marked as ‘PCR product’) that was investigated by bisulphite treatment followed by subclone sequencing and that contained seven differentially methylated CpG sites. The position of the common ∼1.8 kb microdeletion4 associated with BWS is marked with a red bar, and the OCT4 binding site mutation is also shown, as well as the sequencing result of II-3 before and after McrBc digestion of methylated DNA. Please note that the g.2023019A>G mutation corresponds to the T>C mutation on the antisense strand, which was described by10. The position of CTCF binding sites (CTS) in ICR1 are shown, and of these CTS6 and CTS1 were investigated by next generation bisulphite sequencing.
To investigate whether the same tendency could be found in the CTCF binding sites (called CTS and numbered from 1 to 7) of the ICR1 repeats (Figure 2), the degree of methylation of two such sites was investigated by highly quantitative next generation bisulphite sequencing: CTS6 in B-type repeat B1, on the H19-side of ICR1, and CTS1 in B-type repeat B7, on the IGF2-side of ICR1 (Figure 2). Although CTS1 was fully methylated already in the grandmother’s children (II-1, II-3 and II-4; Supplementary Table 1), CTS6 showed the same tendency towards increased methylation from generation II to III. In the mothers of generation II, the average degree of methylation was 73%, and in their children, the average degree of methylation was 81% (Figure 1). It thus seems that CTS1 was more prone to acquire methylation than CTS6.8 To further illustrate the molecular correlation to the observed clinical anticipation, methylation heat maps of the degree of CTS1 and CTS6 methylation from the grandmother to daughters to grandchildren are shown in Figure 3. Of note, the degree of methylation in CTS6 correlates well with the clinical severity (Figure 3), that is, the number of BWS symptoms and findings (see above; II-3

DNA methylation analysis: Heat maps of the methylation patterns obtained by next generation bisulphite sequencing of two CTCF binding sites: CTS1 and CTS6. The heat maps are ordered from left to right by the degree of methylation, and this also corresponds to the clinical severity. Only data from individuals harbouring the allele with the OCT4 binding site mutation is shown. The pedigree marks below the panels are the same as in Figure 1. Lines represent sequence reads, columns CpGs. Blue – unmethylated – maternal (mat); red – methylated – paternal (pat); white – missing sequence information.
As dominant BWS can be associated with mutations in ICR1, between the H19 noncoding RNA gene and the growth factor gene IGF2, this region was examined for mutations or deletions. Long-range PCR did not detect any microdeletions in ICR1 that could explain imprinting disturbances. However, ICR1 sequencing revealed the same maternal point variant NCBI36:11:g.1979595T>C (NCBI37:11:g.2023019T>C, see Figure 2) that had been described previously as a cause of BWS in three brothers.10 To determine the parental origin of the variant, genomic DNA was digested with the methylation-sensitive restriction enzyme McrBC, which digests methylcytosine-containing DNA, and then amplified and sequenced. In the digested DNA from II-1 and II-3, the variant became apparently homozygous (II-3 result shown in Figure 2), indicating that it was present on the partially unmethylated, and therefore maternally inherited allele. In I-2, the non-affected grandmother, the opposite was found, indicating that the point variant was on the grandmother’s paternal allele. Haplotype and sequencing information showed that the three sisters in generation II and the three affected children in generation III inherited this point variant, but not by the unaffected DZ sibling in generation III (Supplementary Figure 2).
Discussion
From a clinical point of view, the second generation tall stature and third generation BWS in our family suggested anticipation (Figure 1). Alternatively, the increased clinical severity might be a random result of variable expression, not uncommon in BWS (see e.g. Scott et al.11). However, the increasing degree of methylation from the unaffected carrier grandmother of a paternal ICR1 variant over the two subsequent generations gave a molecular correlate to the observed anticipation (Figures 1 and 3, Table 1). The increased degree of ICR1 methylation was found both on routine MLPA-based methylation testing and bisulphite subclone sequencing of the CpG island region in the H19 promoter, and bisulphite next generation sequencing of the CTCF binding site in the B1 repeat region of ICR1 (CTS6), but not the more IGF2-proximal CTCF binding site in the B7 repeat (CTS1) (Figures 2 and 3). Upon ICR1 sequencing to find a cause of the apparent anticipation, a previously found variant affecting OCT4 binding was found (Figure 2).10 It was highly unlikely that this represented a founder mutation as the other family was French and the Norwegian family had no known French roots. Nevertheless, to exclude that this could be a founder mutation, Christine Gicquel was most helpful and sent us the SNP information of the ICR1 locus in their family, and when compared to our family, a common haplotype was excluded. We could therefore conclude that the same NCBI37:11:g.2323019T>C (NCBI36:11:1979595T>C) variant had occurred on two different genetic backgrounds.
Recently, a family with another variant reducing OCT4 binding was described.3 This variant 1979624A>C was 31 nt centomeric to the 1979595T>C variant found by Demars et al.10 and us. Of note, in this family there were two affected brothers with prolonged post pubertal growth and final heights above the 99th centiles (204 cm and 208 cm, respectively), which is atypical for BWS patients. They also had renal problems, one had cysts and the other had a Wilms tumour, and neither was markedly asymmetric. This suggests that there may be clinical features that distinguish ICR1 mutation patients from other patients with BWS. None of the reported patients had the characteristic ear creases and growth did not decelerate in their teens, resulting in final statures above the 97.5th centile. If the latter also will hold true for the third generation in our family is yet unknown, as they are all children. Nevertheless, if a child with BWS and ICR1 hypermethylation has a tall statured mother, this should alert to the possibility of dominant inheritance, especially if the child also has Wilms tumour. Absence of ear creases should strengthen this suspicion.
It has previously been shown that the T>C variant in the ICR region affects OCT4/SOX2 binding,10 and our data can be interpreted to imply that the variant interferes with gonadal switching from paternal to maternal imprinting. The same may be true for the A>C variant reported by Poole et al.3 In the latter case, however, classical BWS occurred already in the first generation. One explanation for this discrepancy could be that the A>C variant is more detrimental to OCT4 binding than the T>C variant. Alternatively, the cause could be random variability in clinical expression. Our family suggests, however, that true anticipation as an explanation for increased clinical severity may take place in BWS. This is supported by a recent report on the consequences of ICR1 microdeletions, where at least two of the six families had features compatible with anticipation, described as ‘a kind of epigenetic memory effect’ by the authors.8 Possibly, the phenomenon of anticipation in BWS is not limited to OCT4 binding site mutations, but could be an explanation for non-penetrance in ICR1 deletion families as well. This has implications for genetic counselling of BWS families with ICR1 deletions or mutations. Transmission through the male germline implies a potential BWS risk not necessarily in the next, but in subsequent generations if the mutation is passed through the female germline. Second, transmission of the mutation through the maternal germline may first give rise to BWS in later (third and fourth) generations if the mutation is ‘mild’.
From a more fundamental biological perspective, our findings give clues to how gonadal imprinting switching is regulated. The grandmother inherited a fully methylated paternal allele containing the T>C variant from her father, and a normal demethylated maternal allele from her mother. In the next generation, her three daughters inherited the grandpaternal ICR1 allele (with the T>C variant) and they had methylation levels of around 73%. This implies either that the grandpaternal allele was incompletely demethylated in their mother’s gonads or that the variant allele was completely demethylated in the germline, but had reduced resistance to subsequent postzygotic remethylation. There are experimental data to support both of these suggestions.12 A recent study showed that interference with OCT4 binding in P19 embryonic carcinoma cells rendered the H19 allele partially resistant to demethylation, but also that when partially methylated, further methylation seeding (remethylation) could easily take place.13 Methylation seeding implies that inappropriate methyl groups recruit binding proteins that directly and indirectly promote both histone compaction and further DNA methylation. Moreover, on a more general genomic scale it has been shown that maintenance of imprinting marks in early zygotic development requires not only protection against postfertilisation demethylation, but also protection against somatic remethylation.14 A similar requirement for OCT4/SOX2-binding elements to maintain the maternal demethylated state has been found in the Prader-Willi/Angelman syndrome imprinting centre.15
To explain anticipation, reduced protection against somatic remethylation is not enough. There must also be an element of demethylation resistance in the germline. However, even if the mutation caused only partial ICR1 demethylation to take place in the female germline (grandmother and daughters), one would still expect less ICR1 methylation in third generation (the children with BWS), not more. This remains true even if a susceptibility to somatic remethylation should vary somewhat from individual to individual. Of note, the grandpaternal (normal) allele inherited by III-4 (also confirmed by haplotyping, see Supplementary Figure 2) was completely demethylated in the same maternal gonad that could not demethylate the grandmaternal allele (II-4 in Figure 1). This indicates that mutations interfering with OCT4 binding may have different effects in paternal and maternal inheritance. Apparently, unlike the paternal allele, the maternal allele is incapable of being actively demethylated in the maternal germline—otherwise there would have been no anticipation. Our hypothesis on differential handling of paternal and maternal alleles in the maternal gonads is illustrated in Figure 4.

The imprinting status of the grand-grand-paternal allele after grandmaternal and maternal transmissions. The OCT4 binding site mutation apparently causes incomplete demethylation upon grandmaternal transmission (A), and further methylation upon maternal transmission (B). Of note, further demethylation, as in A, was not seen in B (marked by the X), suggesting that only paternal alleles can be demethylated in the maternal gonads. Blue=methylated (paternal pattern) and red=unmethylated (maternal pattern).
In conclusion, we have both clinical and molecular evidence for the occurrence of anticipation in rare cases of BWS, and our data also indicate that only paternally inherited H19/IGF2 loci are demethylated in the maternal germline.
References
Weksberg R, Shuman C, Beckwith JB : Beckwith-Wiedemann syndrome. Eur J Hum Genet 2010; 18: 8–14.
Choufani S, Shuman C, Weksberg R : Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet 2010; 154C: 343–354.
Poole RL, Leith DJ, Docherty LE et al: Beckwith-Wiedemann syndrome caused by maternally inherited mutation of an OCT-binding motif in the IGF2/H19-imprinting control region, ICR1. Eur J Hum Genet 2012; 20: 240–243.
De Crescenzo A, Coppola F, Falco P et al: A novel microdeletion in the IGF2/H19 imprinting centre region defines a recurrent mutation mechanism in familial Beckwith-Wiedemann syndrome. Eur J Med Genet 2011; 54: e451–e454.
Riccio A, Sparago A, Verde G et al: Inherited and sporadic epimutations at the IGF2-H19 locus in Beckwith-Wiedemann syndrome and Wilms' tumor. Endocr Dev 2009; 14: 1–9.
Demars J, Rossignol S, Netchine I et al: New insights into the pathogenesis of Beckwith-Wiedemann and Silver-Russell syndromes: contribution of small copy number variations to 11p15 imprinting defects. Hum Mutat 2011; 32: 1171–1182.
Prawitt D, Enklaar T, Gartner-Rupprecht B et al: Microdeletion of target sites for insulator protein CTCF in a chromosome 11p15 imprinting center in Beckwith-Wiedemann syndrome and Wilms' tumor. Proc Natl Acad Sci USA 2005; 102: 4085–4090.
Beygo J, Citro V, Sparago A et al: The molecular function and clinical phenotype of partial deletions of the IGF2/H19 imprinting control region depends on the spatial arrangement of the remaining CTCF-binding sites. Hum Mol Genet 2012; 22: 544–557.
Lutsik P, Feuerbach L, Arand J, Lengauer T, Walter J, Bock C : BiQ analyzer HT: locus-specific analysis of DNA methylation by high-throughput bisulfite sequencing. Nucleic Acids Res 2011; 39: W551–W556.
Demars J, Shmela ME, Rossignol S et al: Analysis of the IGF2/H19 imprinting control region uncovers new genetic defects, including mutations of OCT-binding sequences, in patients with 11p15 fetal growth disorders. Hum Mol Genet 2010; 19: 803–814.
Scott RH, Douglas J, Baskcomb L et al: Constitutional 11p15 abnormalities, including heritable imprinting center mutations, cause nonsyndromic Wilms tumor. Nat Genet 2008; 40: 1329–1334.
Hori N, Nakano H, Takeuchi T et al: A dyad oct-binding sequence functions as a maintenance sequence for the unmethylated state within the H19/Igf2-imprinted control region. J Biol Chem 2002; 277: 27960–27967.
Hori N, Yamane M, Kouno K, Sato K : Induction of DNA demethylation depending on two sets of Sox2 and adjacent Oct3/4 binding sites (Sox-Oct motifs) within the mouse H19/insulin-like growth factor 2 (Igf2) imprinted control region. J Biol Chem 2012; 287: 44006–44016.
Proudhon C, Duffie R, Ajjan S et al: Protection against de novo methylation is instrumental in maintaining parent-of-origin methylation inherited from the gametes. Molecular cell 2012; 47: 909–920.
Kaufman Y, Heled M, Perk J, Razin A, Shemer R : Protein-binding elements establish in the oocyte the primary imprint of the Prader-Willi/Angelman syndromes domain. Proc Natl Acad Sci USA 2009; 106: 10242–10247.
Acknowledgements
We are most grateful to the family for giving us the opportunity to gain further insight into mechanisms for BWS and Melanie Heitmann for expert technical assistance. This work was supported by HelseVest project no. 911744 and by the Bundesministerium für Bildung und Forschung (Network Imprinting diseases, grant No. 01GM1114A to K.B.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
This work is licensed under a Creative Commons Attribution 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by/3.0/
About this article
Cite this article
Berland, S., Appelbäck, M., Bruland, O. et al. Evidence for anticipation in Beckwith–Wiedemann syndrome. Eur J Hum Genet 21, 1344–1348 (2013). https://doi.org/10.1038/ejhg.2013.71
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.71
Keywords
This article is cited by
-
Imprinting disorders
Nature Reviews Disease Primers (2023)
-
Search for cis-acting factors and maternal effect variants in Silver-Russell patients with ICR1 hypomethylation and their mothers
European Journal of Human Genetics (2019)
-
Genomic imprinting disorders: lessons on how genome, epigenome and environment interact
Nature Reviews Genetics (2019)
-
The extent of DNA methylation anticipation due to a genetic defect in ICR1 in Beckwith-Wiedemann syndrome
Journal of Human Genetics (2019)
-
Clinical and molecular diagnosis, screening and management of Beckwith–Wiedemann syndrome: an international consensus statement
Nature Reviews Endocrinology (2018)
