
Abstract
Congenital cataracts are an important cause of bilateral visual impairment in infants. Through genome-wide linkage analysis in a four-generation family of Irish descent, the disease-associated gene causing autosomal-dominant congenital nuclear cataract was mapped to chromosome 4p16.1. The maximum logarithm of odds (LOD) score was 2.62 at a recombination fraction θ=0, obtained for marker D4S432 physically close to the Wolfram gene (WFS1). By sequencing the coding regions and intron–exon boundaries of WFS1, we identified a DNA substitution (c.1385A-to-G) in exon 8, causing a missense mutation at codon 462 (E462G) of the Wolframin protein. This is the first report of a mutation in this gene causing an isolated nuclear congenital cataract. These findings suggest that the membrane trafficking protein Wolframin may be important for supporting the developing lens.
Similar content being viewed by others

Introduction
Cataract is an opacification of the lens or the lens capsule in the eye and is the commonest cause of childhood blindness in the world, with an incidence of 1–3 per 10 000 live births.1 During infancy and early childhood without treatment, it frequently results in visual impairment and can lead to irreversible amblyopia. About one-third of congenital cataracts are familial, however, it can occur in isolation or in association with other systemic abnormalities and is also a predominant feature in >200 genetic disorders.2 Inherited congenital cataract is genetically heterogeneous and displays diverse phenotypic features. The phenotypic classification is based on the position and type of the lens opacity including: anterior polar, posterior polar, nuclear, lamellar, coralliform, blue-dot (cerulean), cortical, pulverulent, polymorphic and complete cataract.3
Although non-syndromic cataract has been associated with the three major forms of Mendelian inheritance, most cataracts show autosomal-dominant (AD) inheritance with complete penetrance and variable expressivity. Progress has been made in elucidating cataract-causing mutations in human genes encoding the transparent intracellular lens proteins (crystallins), membrane gap junction proteins (connexins), water channel proteins (aquaporins), solute carrier protein (SLC16A12), various cytoskeletal proteins (for example, phakinin, filensin and vimentin), transcription factors (FOXE3, EYA1, PAX6, MAF and PITX3) and transmembrane proteins (TMEM114, LIM2, CHMP-4B and EPHA2).4 This insight also explains the basis for marked genotypic and phenotypic heterogeneity. Here, we report the first mutation in WFS1 gene causing an isolated AD nuclear cataract with no other ocular or systemic abnormality.
Methods
Phenotyping
A four-generation family of Irish descent with AD cataract was identified from the genetic clinic database at Moorfields Eye Hospital, London, UK. Informed consent was obtained from all participants, consistent with Local Ethics Committee approval. Family members underwent a full ophthalmological examination including visual acuity, slit lamp and retinal examination, tonometry and ultrasonography, paying particular attention to the lens and cataract morphology. No other ocular or systemic features were identified in affected family members. The type of cataract in this family was a nuclear cataract referring to opacification within the embryonal and/or fetal nuclei of the lens.
Genotyping and linkage analysis
Genomic DNA was extracted from peripheral blood lymphocytes using the Nucleon II DNA extraction kit (Scotlab Bioscience, Strathclyde, Scotland, UK).
Genotype data of 16 individuals from this family (Figure 1) were generated using the 50K Array Xba 240 Assay Kit from GeneChip Human Mapping 100k Set (Affymetrix, High Wycombe, UK). Initial checks of the results were performed with GeneChip Command Console Viewer (v1.1.0.845). Genotyping Console (v3.0.2) assigned individual genotypes. Alohomora version 0.30 (Max Delbrück Center for Molecular Medicine, Berlin, Germany) was used to prepare the raw genotype data for total linkage analysis and for PedCheck (version1.1, Jeff O’Connell; University of Pittsburgh, Pittsburgh, PA, USA) to detect and remove Mendelian errors from the data. Genehunter (version 2.1_r5 beta) was used to perform the subsequent parametric linkage analysis with dominant inheritance and full penetrance of the disease allele with a frequency of 0.0001 in the general population.

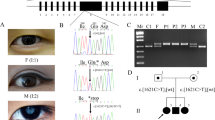
Abridged pedigree of the nuclear cataract family used in this study showing the segregation of five chromosome 4p markers listed in descending order. Squares and circles symbolize males and females, respectively. Open and filled symbols indicate unaffected and affected individuals. The disease haplotype is shown in the box.
The region showing significant logarithm of odds (LOD) score was refined using markers from Marshfield (http://research.marshfieldclinic.org), GDB Human genome database and Ensemble database (http://www.ensembl.org). Genotyping was performed using GeneMapper (version 4.0, Applied biosystems, Warrington, UK) on a ABI PRISM 3730 Genetic Analyzer (Applied Biosystems). Two-point linkage analysis was performed using the MLINK component of the LINKAGE program package version 5.10. A full penetrance and a gene frequency of 0.0001 were used for the cataract locus. The pedigree and haplotype data was managed by Cyrillic software (version 2.1.3).
Sequencing
Agilent SureSelect Human Whole Exon 50 Mb capture kit was used to create the enriched library, which was amplified and then sequenced using a HiSeq2000. A total of 66 million, 100 bp paired-end reads were produced. BWA version 0.5.9 was used to map these reads to the hg19 UCSC reference genome. The GATK variant caller was used following best practices guidelines v3. This included quality score recalibration to normalize base quality score, indel realignment to improve indel detection and variable quality score recalibration, which builds a model for accurate variant detection using publicly available data and ancillary data sets. ANNOVAR was used for variant annotation. A total of 19 726 variants were called with 97.3% being present in dbSNP. The Ti/tv ratio was calculated at 2.31 for all variants. Variants were filtered based on the assumption that the variant of interest is rare within the normal population, to this end dbSNP 135 and 1000genomes (2012 February release) were used. Following this, filtering was done using linkage analysis on chromosome 4p16 region.
Immunoblotting
Total protein from six whole eyes at E18.5, or the lens from two eyes at P3 or P21 were extracted in lysis buffer (50 mM HEPES pH 7.5, 50 mM NaCl, 5 mM EDTA, 1% Triton-X, 10 mM sodium pyrophosphate) that was supplemented with 0.5% phosphatase inhibitor cocktail (Roche Diagnostics, Quebec City, QC, Canada) and 0.5% protease inhibitor cocktail mix (Sigma, Oakville, ON, Canada). Lens homogenates were obtained using short bursts of sonication at 4 °C for 1 min. Insoluble material was removed by centrifugation at 12 000 g for 30 min. Protein concentrations of extracts were determined using a protein assay kit (Pierce, Rockford, IL, USA). Equivalent protein samples and a protein marker (GenScript fluorescent protein marker) were mixed with NuPAGE sample buffer (Invitrogen, Burlington, ON, Canada) and separated by 12% SDS-polyacrylamide gel electrophoresis. Fractionated proteins were transferred to Immobilon-FL membrane (Millipore, Billerica, MA, USA) and then blocked overnight at 4 °C with 5% non-fat milk powder in PBS/0/1% Tween-20 (PBST). Membranes were incubated for 2 h at RT with primary antibodies (1:1000; rabbit polyclonal to human WFS1, Abcam or 1:5000; GAPDH loading control, Abcam, Toronto, ON, Canada) and then washed three times for 15 min each in PBST. Membranes were then incubated in the dark with a Li-COR secondary antibody (IRDye 680LT goat anti-rabbit, Mandel Scientific, Guelph, ON, Canada). After the membrane was washed three times in PBST in the dark, protein bands were visualized using a Li-COR Odyssey detector.
Immunocytochemistry
Mouse embryo heads at E12.5 and E18.5 were dissected and fixed in 4% paraformaldehyde in PBS at 4 °C overnight, cryoprotected in 30% sucrose in PBS at 4 °C. Eyes were frozen in PolyFreeze medium (Polysciences, Warrington, PA, USA) and 10 μm cryosections were transferred to microscope slides. Sections were incubated with blocking buffer (3% goat serum+0.1% Triton-100 in PBS) for 1 h at RT and then incubated overnight at 4 °C with primary rabbit polyclonal antibody to human WFS1 (1:500, Abcam) in blocking buffer. Sections were washed three times for 15 min each with PBS and incubated with secondary goat anti-rabbit antibody conjugated to Alexa-488, diluted 1:1000 in blocking buffer. Fluorescent images were obtained using a Leica MZ16 FA fluorescence stereomicroscope (Meyer Instruments, Houston, TX, USA). Comparative H&E sections were obtained from wax-embedded eyes at the same time points.
Results
A four-generation family with nuclear cataract, comprising 16 members of the pedigree (Figure 1) including 11 affected individuals, 1 unaffected individual and 4 spouses were genotyped with SNP markers using GeneChip Human Mapping 50K Array. Linkage analysis identified a likely disease-haplotype interval on chromosome 4p16 (rs722867-4.6 Mb-rs1401438) and 11p13 (rs2420520-0.45 Mb-rs1396880). Further, microsatellite markers were used to narrow down these regions on chromosomes 4p16 and 11p13. Data from microsatellite markers excluded the region from 11p13 locus (data not shown) and two-point-positive LOD score was obtained only on chromosome 4p16 with marker D4S432 (Z=2.62 at θ=0.00). Recombinant chromosomes were identified with the marker D4S2925 in affected individual III-5 and IV-2 and the marker D4S2949 in unaffected individual IV-4. This 6.6 Mb region of chromosome 4p16.1 encompasses 31 genes. Of these genes, we fully sequenced the coding exons and exon–intron boundaries of OHMX1, CPZ, C4orf23, ACOX3, HTRA3, SH3TC1, DRD5, SLC2A9, WDR1, ZNP, CLNK, H3ST1 and ABLIM2 and no pathogenic changes were identified. To make it cost effective and comprehensive, one of the affected individuals IV-5 was further analyzed by total exome sequencing.
A total of 84 SNPs were found in 6.6 Mb region between markers D4S2925 and D4S2949 of which only one did not appear in the public databases. A single non-synonymous SNV was found that was predicted as damaging/intolerant by both SIFT and POLYPHEN programs within the WFS1 gene, and was not found in the 1000 genome database. As the WFS1 gene has been associated with other eye abnormalities we considered this as a good candidate. To confirm the presence of this, SNV direct genomic sequencing of all the coding exons of WFS1 was undertaken in the whole family. A missense mutation (A-to-G) in exon 8 was identified as causative of the SNV, which cosegregated with disease in the family (Figure 2). This A-to-G transition at nucleotide position 1385 resulted in the substitution of a negatively charged glutamic acid with neutral amino-acid glycine at codon 462 (p.E462G).

Sequence analysis of WFS1 with unaffected individual (upper chromatogram illustrates a normal control) and a missense mutation (c.1385A-to-G; shown in an affected individual).
WFS1 mRNA is known to be expressed in pancreas, brain, heart, skeletal muscle, placenta, lung, liver, kidney, eye5, 6 and the lens (GEO database GSM41475). To confirm the lens RNA expression data, we used a rabbit polyclonal antibody to a 110 amino-acid peptide at the N-terminal region of Wolframin (Abcam ab48169) for western blotting (Figure 3a) and immunocytochemistry (Figures 3b and c). Western blot analysis of whole mouse eye at E18.5 and in whole lens tissue extract at P3 and P21 showed the expected Wfs1 100 kDa band (Figure 3a). Immunocytochemistry demonstrated high-level expression of Wfs1 in the developing mouse lens at E18.5, but not at E12.5 (Figure 3).

Expression of Wfs1 in the developing mouse eye. Left panel (a): western blot for Wfs1 protein. Lane1, whole-eye extract at E18.5; Lane 2, lens extract at P3, Lane3, lens extract at P21. M, GenScript protein marker. Antibody to GAPDH used as loading control. Right panels (b and c): upper images are sectioned stained without primary antibody; middle images are immunolabelling of Wfs1 (white arrows) in the developing mouse eye; lower images are H&E wax sections showing corresponding eye histology. L, lens; R, retina; E, eyelid.
Discussion
In this report, we have performed a total genome search on a four-generation family and mapped a novel locus for nuclear cataract on chromosome 4p16. We have also identified an underlying missense mutation (p.E462G) in the WFS1 gene. The WFS1 gene is comprised of eight exons and encodes an 890 amino-acid Wolframin protein. This mutation is in the cytoplasmic loop linking transmembrane domains 4 and 5. The glutamic acid at position 462 is conserved in the WFS1 protein in human, mouse, chicken, chimpanzee, dog, cow, monkey and rat. This mutation was not found in a panel of 100 normal unrelated white European individuals, excluding the possibility that it may be a polymorphism. We screened a panel of 50 unrelated individuals from families diagnosed with AD congenital cataract and did not find any other mutation in the WFS1 gene.
Wolframin is a hydrophobic and tetrameric protein with nine transmembrane segments and large hydrophilic regions at both termini. Previous studies have demonstrated that Wolframin is an endoplasmic reticulum (ER) membrane-embedded protein.7 ER localization suggests that Wolframin has physiological function in membrane trafficking, secretion, processing and/or regulation of ER calcium homeostasis. Disturbances or overloading of these functions induce ER stress responses, including apoptosis.8 Mutations in WFS1 are known to cause autosomal-recessive Wolfram syndrome.9 Wolfram syndrome (WFS), was first described by Wolfram and Wagener in 1938,10 and is a severe neurodegenerative disease mainly characterized by juvenile-onset diabetes mellitus and optic atrophy (OMIM 222300). The other features of this syndrome include diabetes insipidus, sensorineural hearing loss, peripheral neuropathy, ataxia, psychiatric problems, renal-tract abnormalities, bladder atony and male hypogonadism.11
It has been established that mutations in the WFS1 gene can cause variable expression, giving rise to different clinical complications such as hearing, diabetes, vision and depression.12, 13 WFS1 has been implicated in families with autosomal-dominant isolated low-frequency sensorineural hearing loss.14 Hansen et al15 in 2005 studied seven Danish families with Wolfram syndrome, five families has shown bilateral cataract along with other systemic abnormalities (retarded puberty, diabetic retinopathy, growth retardation), suggesting cataract as a part of the Wolfram syndrome. A missense mutation (E864K) in the WFS1 gene was also reported in a Danish family with autosomal-dominant juvenile-onset optic atrophy in combination with hearing impairment.16
Rendtorff et al17 reported a pair of siblings, followed over 17 years, who manifested congenital or early childhood cataracts, diabetes insipidus, diabetes mellitus, optic atrophy and deafness. Recently WFS was described in a 44-year-old patient with diabetes mellitus, central respiratory failure, cognitive impairment, ataxia and parkinsonism.18 All of these studies have provided the evidence for phenotypic heterogeneity, and that the genotype–phenotype relationships are still not clear. However, it does appear that the majority of recessive WFS1 inactivating mutations cause typical Wolfram syndrome, whereas dominant non-inactivating mutations are associated with less severe, but more varied phenotypic manifestations. In this study, we report a missense mutation (c.A1385G; p.E462G) in WFS1 causing only congenital nuclear cataract.
The Wolframin protein is likely to have several functions, being involved in calcium homeostasis and possibly in other ion transport, as well as a negative regulator of ER stress.19 The induction of ER stress in the lens during development has been shown to cause apoptotic cell death in lens epithelial cells,20 suggesting that the lens would be at risk with defective Wolframin function. Our experiment shows this protein is highly expressed in the developing lens (Figure 3) and likely to be involved in membrane trafficking.
The p.E462G change resides in the cytoplasmic domain and possibly affects the protein–protein interaction relating to the lens. It is likely that different domains perform unique functions in different types of tissues. These findings suggest that the membrane trafficking protein Wolframin might be important for supporting the developing lens. Teasing out various interactions of Wolframin protein in the developing lens will help in the understanding of this unique multifunctional protein and would provide additional benefit for genetic counseling of patients.
References
Reddy MA, Francis PA, Berry V, Bhattacharya SS, Moore AT : Molecular genetic basis of inherited cataract and associated phenotypes. Surv Ophthalmol 2004; 49: 300–315.
Krumpaszky HG, Klauss V : Epidemiology of blindness and eye disease. Ophthalmologica 1996; 210: 1–84.
Francis PJ, Berry V, Bhattacharya SS, Moore AT : The genetics of childhood cataract. J Med Genet 2000a; 37: 481–488.
Shiels A, Bennett TM, Hejtmancik JF : Cat-Map: putting cataract on the map. Mol Vis 2010; 16: 2007–2015.
Schmidt-Kastner R, Kreczmanski P, Preising M et al: Expression of the diabetes risk gene wolframin (WFS1) in the human retina. Exp Eye Res 2009; 89: 568–574.
Kawano J, Tanizawa Y, Shinoda K : Wolfram syndrome 1 (Wfs1) gene expression in the normal mouse visual system. J.Comp Neurol 2008; 510: 1–23.
Takeda K, Inoue H, Tanizawa Y, Matsuzaki Y et al: WFS1 (Wolfram syndrome 1) gene product: predominant subcellular localization to endoplasmic reticulum in cultured cells and neuronal expression in rat brain. Hum Mol Genet 2001; 10: 477–484.
Hofmann S, Philbrook C, Gerbitz KD, Bauer MF : Wolfram syndrome: structural and functional analyses of mutant and wild-type Wolframin, the WFS1 gene product. Hum Mol Genet 2003; 12: 2003–2012.
Strom TM, Hörtnagel K, Hofmann S et al: Diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD) caused by mutations in a novel gene (Wolframin) coding for a predicted transmembrane protein. Hum Mol Genet 1998; 7: 2021–2028.
Wolfram DJ, Wagener HP : Diabetes mellitus and simple optic atrophy among siblings: report of four cases. Mayo Clin Proc 1938; 13: 715–718.
Barrett TG, Bundey SE, Macleod AF : Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet 1995; 346: 1458–1463.
Cryns K, Sivakumaran TA, Van den Ouweland JM et al: Mutational spectrum of the WFS1 gene in Wolfram syndrome, nonsyndromic hearing impairment, diabetes mellitus, and psychiatric disease. Hum Mutation 2003; 22: 275–287.
Tranebjaerg L, Barrett T, Rendtorff ND : WFS1-related disorders. GeneReviews at GeneTests: Medical Genetics Information Resource. University of Washington: Seattle, 2009, pp 1997–2008.
Young TL, Ives E, Lynch E et al: Non-syndromic progressive hearing loss DFNA38 is caused by heterozygous missense mutation in the Wolfram syndrome gene WFS1. Hum Mol Genet 2001; 10: 2509–2514.
Hansen L, Eiberg H, Barrett T et al: Mutation analysis of the WFS1 gene in seven Danish Wolfram syndrome families; four new mutations identified. Eur J Hum Genet 2005; 12: 1275–1284.
Eiberg H, Hansen L, Kjer B et al: Autosomal dominant optic atrophy associated with hearing impairment and impaired glucose regulation caused by a missense mutation in the WFS1 gene. J Med Genet 2006; 43: 435–440.
Rendtorff ND, Lodahl M, Boulahbel H et al: Identification of p.A684V missense mutation in the WFS1 gene as a frequent cause of autosomal dominant optic atrophy and hearing impairment. Am J Med Genet A 2011; 6: 1298–1313.
Waschbisch A, Volbers B, Struffert T et al: Primary diagnosis of Wolfram syndrome in an adult patient-case report and description of a novel pathogenic mutation. J Neurol Sci 2011; 300: 191–193.
Rigoli L, Lombardo F, Di Bella C : Wolfram syndrome and WFS1 gene. Clin Genet 2011; 79: 103–117.
Pyati UJ, Gjini E, Carbonneau S et al: Look AT. p63 mediates an apoptotic response to pharmacological and disease-related ER stress in the developing epidermis. Dev Cell 2011; 21: 492–505.
Acknowledgements
This work was supported by The Wellcome Trust project grant 063969/Z/01, EU project ‘PYTHIA’ (FP7-ICT2–224030), and ‘the National Institute for Health Research (NIHR) Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.’ We would like to thank the members of the family for taking part in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Electronic-database information, The accession number and URL for data in this article are as follows
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim (for WFS1 (MIM 606201)) Marshfield, GDB Human genome database (http://research.marshfieldclinic.org) and ENSEMBLE genome data resources (http://www.ensembl.org).
Rights and permissions
About this article
Cite this article
Berry, V., Gregory-Evans, C., Emmett, W. et al. Wolfram gene (WFS1) mutation causes autosomal dominant congenital nuclear cataract in humans. Eur J Hum Genet 21, 1356–1360 (2013). https://doi.org/10.1038/ejhg.2013.52
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.52
Keywords
This article is cited by
-
Wfs1E864K knock-in mice illuminate the fundamental role of Wfs1 in endocochlear potential production
Cell Death & Disease (2023)
-
The genetic and clinical characteristics of WFS1 related diabetes in Chinese early onset type 2 diabetes
Scientific Reports (2023)
-
Identification of novel variants in Turkish families with non-syndromic congenital cataracts using whole-exome sequencing
International Ophthalmology (2023)
-
A novel mutation of WFS1 gene leading to increase ER stress and cell apoptosis is associated an autosomal dominant form of Wolfram syndrome type 1
BMC Endocrine Disorders (2021)
-
Genetic landscape of isolated pediatric cataracts: extreme heterogeneity and variable inheritance patterns within genes
Human Genetics (2019)
