
Abstract
Autosomal dominant sensorineural hearing loss (ADSNHL) is extremely genetically heterogeneous, making it difficult to molecularly diagnose. We identified a multiplex (n=28 affected) family from the genetic isolate of Newfoundland, Canada with variable SNHL and used a targeted sequencing approach based on population-specific alleles in WFS1, TMPRSS3 and PCDH15; recurrent mutations in GJB2 and GJB6; and frequently mutated exons of KCNQ4, COCH and TECTA. We identified a novel, in-frame deletion (c.806_808delCCT: p.S269del) in the voltage-gated potassium channel KCNQ4 (DFNA2), which in silico modeling predicts to disrupt multimerization of KCNQ4 subunits. Surprisingly, 10/23 deaf relatives are non-carriers of p.S269del. Further molecular characterization of the DFNA2 locus in deletion carriers ruled out the possibility of a pathogenic mutation other than p.S269del at the DFNA2A/B locus and linkage analysis showed significant linkage to DFNA2 (maximum LOD=3.3). Further support of genetic heterogeneity in family 2071 was revealed by comparisons of audio profiles between p.S269del carriers and non-carriers suggesting additional and as yet unknown etiologies. We discuss the serious implications that genetic heterogeneity, in this case observed within a single family, has on molecular diagnostics and genetic counseling.
Similar content being viewed by others


Introduction
Sensorineural hearing loss (SNHL) is a common condition and is extremely genetically heterogeneous (>100 mapped loci and >60 genes; http://hereditaryhearingloss.org) with limited genotype–phenotype correlations,1 which poses a significant challenge to diagnose via molecular strategies. For example, the majority of autosomal dominant (AD) cases are characterized by high-frequency loss because of both known and novel mutations in any one of 26 genes (http://hereditaryhearingloss.org; accessed 13 July 2012). For dominant disorders, the inheritance patterns do not always follow classic Mendelian patterns because of one or more well-recognized phenomena of reduced penetrance, variable expressivity and the presence of phenocopies. Phenocopies are a serious concern when a single diagnosis can result in a failed attempt to discover the diagnosis for multiplex families, especially in the case of whole-exome/genome approaches.
In the course of a project to identify the molecular etiology of SNHL in the Newfoundland population, we previously identified several deafness genes segregating in extensive pedigrees including WFS1,2 TMPRSS33 and PCDH15.4 Herein we report on a multiplex (n=28 affected) family of English extraction identified with a novel in-frame deletion within KCNQ4 (DFNA2) (p.S269del). This was carried out using a step-wise strategy based on population-specific deafness alleles and frequently mutated autosomal dominant SNHL (ADSNHL) genes.5 This is the first phosphate-binding loop (P-loop) deletion described in KCNQ4. Significant genetic heterogeneity within this single family has broad implications for molecular diagnosis and genetic counseling in families with ADSNHL.
Subjects and methods
Clinical recruitment and diagnosis
This project is part of a large study of hereditary hearing loss in the province of Newfoundland and Labrador, Canada. We ascertained a six generation Newfoundland family with a variable pattern of SNHL segregating as an AD trait (Figure 1) with surnames suggestive of English origin.6 Informed consent, family history and permission to access medical records and audiograms were obtained as per approved protocol #01.186 (Human Research Ethics Board, St John’s, NL, Canada). All phenotypic data were reviewed by audiologists (AG & SGS) and classified according to the recommendations of the GENDEAF study group (http://hereditaryhearingloss.org). Audiometric thresholds reported are the measured air conduction thresholds. Audiometric configuration, symmetry, severity and age of onset were assessed in all affected members with hearing loss.

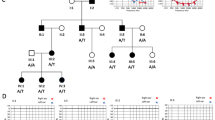
Full, six-generation pedigree of family 2071, a Caucasian family of Northern European descent with a variable form of SNHL in an apparent AD pattern. Family members with SNHL are shaded black. Polymorphic microsatellite markers and SNPs spanning 6.2 Mb in the vicinity of KCNQ4 are given on the left and alleles (size in base pairs or specific bases) given on the right. The disease-associated KCNQ4 mutation, p.S269del, is noted as del (deletion) and the normal sequence as wt (wild type). The pedigree was drawn using Progeny Pedigree Drawing Software (Progeny Software, LLC, Delray Beach, FL, USA).The yellow haplotype indicates the disease-associated haplotype, and genotypes within brackets are inferred.
Candidate gene sequencing
Genomic DNA was extracted from peripheral leukocytes of all available family members according to standard established methods.7 Genomic DNA from the proband (Figure 1; IV-11) was screened by Sanger sequencing for population-specific mutations in WFS1, TMPRSS3 and PCHD15, and common recurrent mutations in GJB2 and GJB6. This was followed by targeted sequencing of exons 5–7 in KCNQ4, exons 4, 5, 12 in COCH and exons 5, 9–14, 17, 18, 20 in TECTA based on their association with bilateral symmetrical, sloping, SNHL,5 as was seen in the proband and the majority of first- and second-degree relatives of the proband (Figure 3). To check for other potential pathogenic mutations in the DFNA2 locus, we fully sequenced the remaining exons of KCNQ4 and the entirety of GJB3 (DFNA2B) in selected family members. Variants of interest were subjected to cascade sequencing (affected n=23; unaffected n=19) and allele frequencies determined using ethnically matched population controls from Newfoundland. Sanger sequencing was carried out on an ABI PRISM 3130XL DNA Analyzer (Applied Biosystems, Foster City, CA, USA) as previously described.8 To identify other candidate genes to screen, audiograms from selected subjects were submitted to Audiogene (http://audiogene.eng.uiowa.edu/audiogram) for computerized comparison with known average audiograms of 16 AD loci.
Microsatellite and haplotype analysis
Microsatellites were genotyped according to standard procedures. All available family members were genotyped for DFNA2A/B-linked markers (see Supplementary Table 1) and alleles were scored with GeneMapper software (v.4.0; Applied Biosystems). Haplotypes were manually constructed (PCR primers and conditions available on request).
Linkage analysis
Linkage calculations were performed using the Linkage Program MLINK, version 5.1.9 Two-point linkage analysis was carried out using MLINK on four microsatellite markers (D1S195, D1S472, D1S2706 and D1S2130). The frequency of hearing loss because of mutations in DFNA2A was set at 0.001 and penetrance set at 100%. For each marker allele frequencies were set equal at 1/N.
Structural modeling of KCNQ4
The structure of KCNQ4 (residues 247–329 wild-type and serine deleted) was modeled using the Swiss protein data bank (http://swissmodel.expasy.org/repository/). The resolved NMR structure of WSK3 (pdb ID: 2k1eB), a water soluble analog of the potassium ion channel KcsA10 was chosen as the best fit template by the database and lowest energy state predictions were generated. Analysis of predicted models and generation of ribbon and stick models were performed using PyMOL v1.30 (Schrodinger, Cambridge, MA, USA) (Figure 2d).

Observed and predicted changes to KCNQ4. (a) Electropherogram of genomic DNA from proband (IV-11). Mutation surveyor readout (top to bottom): KCNQ4 reference sequence (NM_004700); proband sample; conserved allele; mutant allele; mutant allele with 3-bp deletion. (b) Topology of DFNA2 deletion mutations in the KCNQ4 channel protein showing the novel in-frame 3-bp deletion (p.S269del) identified in family 2071 within the conserved P-loop domain between S5 and S6. (c) Conservation of the KCNQ4 protein using ClustalW analysis and WebLogo display. Homo sapiens (NP_004691), Pan troglodytes (XP_513360), Canis luparis familiaris (XP_539568), Rattus norvegicus (XP_233477.4), Mus musculus (NP_001074611.1), Gallus gallus (ABC75089) and Xenopus tropicalis (NP_001072924). (d) Three-dimensional schematic of the area surrounding the P-loop of the KCNQ4 protein. The structure on the left represents the normal protein structure and on the right is the predicted mutant KCNQ4 protein with absence of the S269 residue. The alteration in the three-dimensional positioning of the F267 residue causes steric hinderance to the formation of subunits of KCNQ4 into a functional channel.
Results
Candidate gene sequencing
In the initial phase of the project, sequence analysis of the index case (proband) for population-specific alleles in PCDH15, WFS1 and TMPRSS3 yielded no positive hits. Targeted sequencing of exons 5–7 in KCNQ4, exons 4, 5, 12 in COCH, and exons 5, 9–14, 17, 18, 20 in TECTA revealed that the proband with bilateral SNHL is heterozygous for a novel 3 base-pair deletion, c.806_808delCCT, in exon 4 of KCNQ4. KCNQ4 is a known deafness gene that encodes a potassium channel expressed in the inner and outer ear, and the P-loop is involved in the formation of the tetrameric structure and function of KCNQ4 channels (Figures 2a and c).11 The c.806_808delCCT mutation predicts the in-frame deletion of serine at position 269 (p.S269del) within the conserved N-terminal region of the P-loop of KCNQ4. Twelve other deaf relatives were also heterozygous for the p.S269del variant, which was absent in all unaffected relatives (Figure 1). Furthermore, c.806_808delCCT was not reported in dbSNP and was absent in 180 ethnically matched Newfoundland control chromosomes.
We modeled the structure of the portions of the KCNQ4 protein containing wild type and S269 deleted KCNQ4 (residues 247–329) based on the resolved NMR structure of the ion channel WSK3. This portion contains the two alpha helices S5 and S6 and their connecting loop. We found several features that may potentially be informative; first, the loop contained a short alpha helix structure that was present in both the wild type and p.S269del, starting a few residues downstream of S269. Thus, this feature does not explain the difference between wild type and p.S269del. Second, we noted a phenylalanine residue (F267) within the loop motif with the side pointed toward the interior of the wild-type model structure, but flipped toward the exterior in the p.S269del model structure. The F267 residue in this external position is predicted to interfere with the multimerization of KCNQ4 subunits (Figure 2d).
After detection of the novel p.S269del mutation in the index patient and mutational analysis on the extended kindred, it became apparent that numerous individuals homozygous for the wild-type allele also presented with SNHL (Figure 1). The affected individuals that did not inherit the KCNQ4 p.S269del variant included three close relatives of the proband (III-3, IV-8 and V-7) and seven more distant relatives (III-5, IV-14, IV-15, IV-18, IV-20, IV-21 and IV-22) representing the entire right-sided branch of the family. Two-point linkage analysis between DFNA2-linked markers and the SNHL trait yielded highly negative LOD scores at all markers when all affected individuals of the left side of the pedigree were considered (Table 2a). Close examination of the audio profiles across the pedigree showed that the deletion carriers consistently presented with bilateral, sloping SNHL whereas the non-deletion carriers presented with SNHL of variable symmetry and configuration (Figures 3 and 4). Therefore, we hypothesized that perhaps more than one gene co-segregates with SNHL in this family and reanalyzed the genotype data. Exclusion of non-deletion carriers from the linkage analysis (changing the affection status from affected to unknown) increased the LOD score across all markers to a maximum of 3.3 at marker D1S2130 (Table 2b). This approach yielded statistically significant evidence for DFNA2-linked SNHL on the left-hand side of the pedigree.

Audiograms of affected family 2071 individuals (left side). (a) Audiograms from members from the left-hand side of family 2071 (Figure 1) with documented SNHL. Select audiograms for affected members who inherited the P-loop deletion (IV-4, IV-6, IV-11, V-2, V-4, V-6 and VI-1). Thresholds for subject VI-I left ear at age 5 are actually bone conduction thresholds as air conduction thresholds were affected by temporary conductive loss unrelated but additional to the sensorineural loss. (b) Select audiograms for affected individuals without the deletion (III-3, IV-8 and V-7). Subjects IV-6, IV-4, IV-11, V-2, V-4, V-6 and VI-1 each show two separate audiograms measured at different ages.

Audiograms of selected deaf individuals from the unsolved branch of family 2071 (right-hand side of Figure 1) showing wide variations in configuration, slope and symmetry of hearing loss.
Given the linkage support of DFNA2 for the portion of the family most closely related to the proband, we investigated the possibility that the p.S269del variant was not disease causing, but was in linkage disequilibrium with a putative pathogenic mutation. The DFNA2 locus harbors two known deafness genes, KCNQ4 (DFNA2A) and GJB3 (DFNA2B). Full bidirectional sequencing KCNQ4 and GJB3 revealed no new variants. Haplotype analysis indicated that the 10 relatives heterozygous for the p.S269del variant shared all alleles spanning a 6.2 Mb region on chromosome 1 between markers D1S195 and D1S2130. This haplotype constellation provided evidence to support a disease-associated DFNA2 haplotype. However, deaf relatives wild type for KCNQ4 did not share a DFNA2 disease haplotype, ruling out the likelihood that the putative mutation was in a non-coding region (or involved an indel not readily detected by Sanger sequencing). The fact that 10 of the 23 relatives of this extended family are deaf suggests that a distinct genetic defect may be causing hearing loss in these patients.
Comparison of phenotype based on carrier status
Phenotype of p.S269del carriers
The configuration of hearing loss in carriers of p.S269del was uniformly bilateral sloping sensorineural loss. While not meeting GENDEAF criteria for progressive loss, severity of loss increased across the life span, as older subjects (IV-11, IV-6 and IV-4) showed more severe hearing loss across all frequencies (Figure 3a). Audiograms of nine subjects (IV-4, IV-6, IV-11, V-1, V-2, V-4, V-5, V-6 and VI-1) showed symmetrical hearing loss, while two subjects (III-2, V-9) showed asymmetrical loss (Table 1). A verbal report of behavioral observation audiometry results of the youngest subject (VI-1) at 21 months indicated possible pre-lingual onset of slight hearing loss. This subject's data (at 5 and 17 years) indicated mild loss confined to high frequencies initially, and subsequent deterioration in all frequencies (Figure 3a).
Phenotype of non-deletion carriers
In contrast, the configuration of hearing loss in patients wild type at DFNA2 was variable and distinct from KCNQ4 p.S269del carriers (Figures 3b and 4). The proband’s 58 year old brother (IV-8) has asymmetrical loss, confined to the high frequencies on the right with slight decline of right hearing over time, and sloping moderate loss on the left progressing to severe flat loss within a 10 year period. The proband’s daughter (V-7) at age 29, shows unilateral left mild, flat, low-frequency SNHL, progressing to moderate, low-frequency ascending hearing loss by age 44, while normal hearing is preserved in the right ear (Figure 3b). Two successive audiograms of the proband’s uncle (III-3) show bilateral symmetrical sloping configuration of hearing loss (Figure 3b), but the degree of severity at age 83 is considerably less than hearing loss severity of three deletion carriers at age 62 (IV-6, IV-4 and IV-11).
Audiograms from all deaf family members were uploaded to Audiogene for computerized comparison with known average audiograms of 16 AD loci. All audio profiles of KCNQ4 p.S269del carriers matched average audiograms for DFNA2. In contrast, matches were found for several relatives on the right-hand side of the pedigree and included DFNA9 (COCH), DFNA15 (POU4F3) and DFNA22 (MYO6). Full Sanger sequencing of MYO6, COCH and POU4F3 in deaf subjects IV-14 and IV-15 yielded no mutations. The non-DFNA2 audio profiles in non-deletion carriers support a distinct and unknown etiology in these patients.
Discussion
We report the first deletion in the P-loop domain of KCNQ4 causing ADSNHL in an extended family of northern European descent from England. The DFNA2 locus was originally mapped and KCNQ4 identified as the causative gene in two ADSNHL families of Northern European descent.12, 13 Potassium channels formed by KCNQ4 have a crucial role in the regulation of neuronal and cochlear receptor excitability, including the inner and outer hair cells of the cochlea where KCNQ4 is expressed.12, 14 The novel p.S269del mutation is predicted to cause the removal of a serine residue between the S5 and S6 transmembrane domains that form the channel pore, which contains a filter that selects for the passage of potassium ions in the inner ear.15 To date, 16 mutations in KCNQ4 are associated with ADSNHL, including 11 missense,11, 12, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 1 nonsense,15 1 splice-site mutation21 and 3 deletions.17, 19, 20 Of the 11 missense mutations reported, 6 are found in the P-loop and cause early-onset all-frequency hearing loss.
To examine the potential effects of the p.S269del variant on the structure of the KCNQ4 protein we carried out in silico protein modeling. Compared with the wild-type KCNQ4 protein, the loss of serine at position 269 is predicted to disrupt a proximal residue, phenylalanine at position 267 (F267), to interfere with the multimerization of KCNQ4 subunits. We postulate that the loop portion of KCNQ4 may act as a multimerization interface and that this difference may cause steric hindrance, disrupting multimerization. KCNQ4 channels are assemblies comprising four protein subunits. Mutations in a single unit can disrupt channel function of the tetramer in a dominant-negative manner, which can be explained by the structure of the channel. If both normal and aberrant alleles are synthesized at the same level and assembled in a stochastic manner, the majority of tetrameric channels would be non-functional as they would contain 1 or more mutant subunits; only 1 out of 16 channels would have 4 wild-type subunits. This would cause heterozygous p.S269del carriers to exhibit a strong dominant-negative effect.
The p.S269del mutation in KCNQ4 was found in a multiplex family of English extraction and represents the fourth KCNQ4 deletion reported and the only deletion discovered inside the P-loop region. The first described deletion, c.211del13, was found in a Belgian family and results in a frameshift after the G70 residue leading to a premature stop codon at amino-acid position 134.17 The second KCNQ4 deletion, c.211delC, was found in a Japanese family and is predicted to generate a truncated protein before the first transmembrane domain.20 Generally, missense mutations in KCNQ4 have been associated with more severe, earlier-onset, all-frequency SNHL, while deletions are reported to cause a milder phenotype characterized by a later age of onset affecting only high frequencies.12, 15, 19, 20, 22 Recently, a third deletion, c.664del18, reported in a Korean family causes the deletion of six amino acids in the intra-membrane loop between the fourth and fifth (S4 and S5) transmembrane domains.19 Baek et al (2010) verified the importance of the S4–S5 loop for proper potassium channel function.
Identification of phenocopies
Surprisingly, both molecular and phenotypic analyses suggest that there are at least two different genes involved in family 2071. The pattern of hearing loss in p.S269del carriers is typical of the general pattern found in DFNA2 cases, which is bilateral, sloping, mild to moderate sensorineural loss, of early onset. Carriers of the p.S269del mutation also show that severity of the loss increases with age across all frequencies.12, 15, 19, 20, 22, 25 All family members heterozygous for the p.S269del mutation were diagnosed with SNHL, indicating that this KCNQ4 mutation is highly penetrant. However, three first-degree relatives of the index case did not inherit the KCNQ4 deletion or the deafness-associated DFNA2 haplotype but inherited the normal chromosome from the parent. As well, neither the P-loop deletion nor the DFNA2 disease-associated haplotype segregated with SNHL on the right-hand side of the pedigree. In fact, two-point linkage analysis yielded highly negative LOD scores for all DFNA2 markers (Table 2a). Comparison of audio profiles of non-deletion carriers revealed different audiometric profiles from that of the deletion carriers, and most likely represents phenocopies.
Audiological/clinical management
The clinical impact of carrying a KCNQ4 mutation is significant for children in the critical early years of language acquisition, and continuing throughout the years of formal education and social development. Audiological data from the youngest subject (VI-1) indicate onset may first affect high frequencies to a mild degree early within the first decade, possibly pre-lingually, with subsequent deterioration in all frequencies. Hearing loss affecting high frequencies has been found in many young school-aged children who carry mutations in KCNQ4, most recently in a 1-year old with a missense mutation in the P-loop.18 Taken together, the evidence points to very early, possibly pre-lingual initiation of HL, a finding of clinical significance. The relatively mild initial loss appearing in the first years of life may escape early detection and opportunity for timely intervention. Sloping hearing loss, which affects high frequencies more severely than low frequencies, causes impaired ability to detect many consonants and results in difficulty hearing speech clearly. This can have significant implications for educational and social success if not addressed through rehabilitative support. Hearing aid technology that provides additional volume equalized to the threshold slope is necessary to improve hearing function, and requires adjustment as the loss continues to develop in all frequencies in order for the wearer to achieve maximum rehabilitative benefit. Identifying extended multiplex families provides natural history information to help determine the age of onset and early patterns of progression for hearing loss. Family 2071 members have benefitted from the use of hearing aids, but none have received a cochlear implant. If hearing deteriorates beyond audible limits, a cochlear implant may be recommended. Further knowledge of the natural history and specific auditory defects caused by this novel KCNQ4 mutation throughout the lifespan could be used to inform selection of the most effective communication strategies and technology.
Serious pitfalls for AD disorders
The molecular diagnosis of family 2071 was confounded by the overwhelming presence of phenocopies, likely due to at least a second unidentified gene. Although the vertical transmission of SNHL was consistent with AD inheritance, including male-to-male transmission (Figure 1), cascade sequencing of the extended family yielded 10 deaf family members wild type at the DFNA2 locus, including three close relatives. At this point, we may have ruled out KCNQ4 p.S269del as the cause of ADSNHL in family 2071, given the variant was not previously identified as pathogenic. However, we also recognized that this family may represent an extreme case of genetic heterogeneity, and explored linkage to DFNA2 in p.S269del carriers on the left-hand side (Figure 1 and Table 2). We also ruled out other putative mutations at DFNA2A/B by full gene sequencing and haplotype analysis of a 6.2 Mb region on chromosome 1 (Figure 1). Comparison of audiograms showed distinct differences in configuration and severity between p.S269del carriers and non-carriers. This finding implies a distinct etiology, likely genetic, on the right-hand side of family 2071. In the sibship containing affected individuals IV-14, IV-15 and IV-18 (Figure 1), only 3 of 15 siblings are affected (20%), and the 3 sibships containing IV-20, IV-21 and IV-22 only contain one affected individual (10%, 11.1% and 14.3% respectively). This low number of affected individuals is compatible with AD disease with reduced penetrance, or autosomal recessive inheritance, or could also be due to chance. Recessive inheritance, particularly pseudodominance, might also be a possibility, given that Newfoundland is a genetic isolate with high inbreeding coefficient and low immigration comparable to the Hutterites or Amish populations.26 To further examine the right-hand side of the family, we will explore whole-genome approaches and alternative models of inheritance.
References
Raviv D, Dror AA, Avraham KB : Hearing loss: a common disorder caused by many rare alleles. Ann N Y Acad Sci 2010; 1214: 168–179.
Young TL, Ives E, Lynch E et al: Non-syndromic progressive hearing loss DFNA38 is caused by heterozygous missense mutation in the Wolfram syndrome gene WFS1. Hum Mol Genet 2001; 10: 2509–2514.
Ahmed ZM, Li XC, Powell SD et al: Characterization of a new full length TMPRSS3 isoform and identification of mutant alleles responsible for nonsyndromic recessive deafness in Newfoundland and Pakistan. BMC Med Genet 2004; 5: 24.
Doucette L, Merner ND, Cooke S et al: Profound, prelingual nonsyndromic deafness maps to chromosome 10q21 and is caused by a novel missense mutation in the Usher syndrome type IF gene PCDH15. Eur J Hum Genet 2009; 17: 554–564.
Hilgert N, Smith RJ, Van Camp G : Forty-six genes causing nonsyndromic hearing impairment: which ones should be analyzed in DNA diagnostics? Mutat Res 2009; 681: 189–196.
Seary ER, Lynch SMP : Family Names of the Island of Newfoundland, Corrected edn St John’s, Newfoundland and Labrador, Canada: Memorial University of Newfoundland, 1998.
Miller SA, Dykes DD, Polesky HF : A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
Merner ND, Hodgkinson KA, Haywood AF et al: Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet 2008; 82: 809–821.
Cottingham RW, Idury RM, Schaffer AA : Faster sequential genetic linkage computations. Am J Hum Genet 1993; 53: 252–263.
Ma D, Tillman TS, Tang P et al: NMR studies of a channel protein without membranes: structure and dynamics of water-solubilized KcsA. Proc Natl Acad Sci USA 2008; 105: 16537–16542.
Mencia A, Gonzalez-Nieto D, Modamio-Hoybjor S et al: A novel KCNQ4 pore-region mutation (p.G296S) causes deafness by impairing cell-surface channel expression. Hum Genet 2008; 123: 41–53.
Kubisch C, Schroeder BC, Friedrich T et al: KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 1999; 96: 437–446.
Coucke P, Van Camp G, Djoyodiharjo B et al: Linkage of autosomal dominant hearing loss to the short arm of chromosome 1 in two families. N Engl J Med 1994; 331: 425–431.
Kharkovets T, Dedek K, Maier H et al: Mice with altered KCNQ4 K+ channels implicate sensory outer hair cells in human progressive deafness. EMBO J 2006; 25: 642–652.
Hildebrand MS, Tack D, McMordie SJ et al: Audioprofile-directed screening identifies novel mutations in KCNQ4 causing hearing loss at the DFNA2 locus. Genet Med 2008; 10: 797–804.
Talebizadeh Z, Kelley PM, Askew JW, Beisel KW, Smith SD : Novel mutation in the KCNQ4 gene in a large kindred with dominant progressive hearing loss. Hum Mutat 1999; 14: 493–501.
Coucke PJ, Van Hauwe P, Kelley PM et al: Mutations in the KCNQ4 gene are responsible for autosomal dominant deafness in four DFNA2 families. Hum Mol Genet 1999; 8: 1321–1328.
Arnett J, Emery SB, Kim TB et al: Autosomal dominant progressive sensorineural hearing loss due to a novel mutation in the KCNQ4 gene. Arch Otolaryngol Head Neck Surg 2011; 137: 54–59.
Baek JI, Park HJ, Park K et al: Pathogenic effects of a novel mutation (c.664_681del) in KCNQ4 channels associated with auditory pathology. Biochim Biophys Acta 2010; 1812: 536–543.
Kamada F, Kure S, Kudo T et al: A novel KCNQ4 one-base deletion in a large pedigree with hearing loss: implication for the genotype-phenotype correlation. J Hum Genet 2006; 51: 455–460.
Su CC, Yang JJ, Shieh JC, Su MC, Li SY : Identification of novel mutations in the KCNQ4 gene of patients with nonsyndromic deafness from Taiwan. Audiol Neurootol 2007; 12: 20–26.
Topsakal V, Pennings RJ, te Brinke H et al: Phenotype determination guides swift genotyping of a DFNA2/KCNQ4 family with a hot spot mutation (W276S). Otol Neurotol 2005; 26: 52–58.
Van Hauwe P, Coucke PJ, Ensink RJ, Huygen P, Cremers CW, Van Camp G : Mutations in the KCNQ4 K+ channel gene, responsible for autosomal dominant hearing loss, cluster in the channel pore region. Am J Med Genet 2000; 93: 184–187.
del Castillo I, Villamar M, Moreno-Pelayo MA et al: A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med 2002; 346: 243–249.
Smith RJH, Hildebrand M : DFNA2 Nonsyndromic Hearing Loss, in Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, (eds) Seattle, WA, USA: University of Washington, 2011.
Rahman P, Jones A, Curtis J et al: The Newfoundland population: a unique resource for genetic investigation of complex diseases. Hum Mol Genet 2003; 12: (Spec No 2): R167–R172.
Acknowledgements
We thank family members for participating and Drs S Kirby, T Batten and B Fernandez for help with clinical recruitment. This work was supported by grants from the Canadian Institutes of Health Research (CIHR MOP-66974), the Canadian Foundation for Innovation (New Investigator Award no. 9384; Leaders Opportunity Fund no.13120) and Genome Canada (Atlantic Medical Genetics and Genomics Initiative) to TLY. We gratefully acknowledge financial support from the Janeway Children’s Hospital Foundation, Memorial University IRIF and the Government of Newfoundland and Labrador. We also thank the Canadian Hard of Hearing Association-NL chapter for their continued support. TLY is a CIHR salary awardee; NA is a CIHR-RPP fellowship awardee.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Abdelfatah, N., McComiskey, D., Doucette, L. et al. Identification of a novel in-frame deletion in KCNQ4 (DFNA2A) and evidence of multiple phenocopies of unknown origin in a family with ADSNHL. Eur J Hum Genet 21, 1112–1119 (2013). https://doi.org/10.1038/ejhg.2013.5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.5
Keywords
This article is cited by
-
Novel KCNQ4 variants in different functional domains confer genotype- and mechanism-based therapeutics in patients with nonsyndromic hearing loss
Experimental & Molecular Medicine (2021)
-
A novel pore-region mutation, c.887G > A (p.G296D) in KCNQ4, causing hearing loss in a Chinese family with autosomal dominant non-syndromic deafness 2
BMC Medical Genetics (2017)
-
A common variant in CLDN14 causes precipitous, prelingual sensorineural hearing loss in multiple families due to founder effect
Human Genetics (2017)
