
Abstract
β-Thalassaemia is one of the most common autosomal recessive single-gene disorder worldwide, with a carrier frequency of 12% in Cyprus. Prenatal tests for at risk pregnancies use invasive methods and development of a non-invasive prenatal diagnostic (NIPD) method is of paramount importance to prevent unnecessary risks inherent to invasive methods. Here, we describe such a method by assessing a modified version of next generation sequencing (NGS) using the Illumina platform, called ‘targeted sequencing’, based on the detection of paternally inherited fetal alleles in maternal plasma. We selected four single-nucleotide polymorphisms (SNPs) located in the β-globin locus with a high degree of heterozygosity in the Cypriot population. Spiked genomic samples were used to determine the specificity of the platform. We could detect the minor alleles in the expected ratio, showing the specificity of the platform. We then developed a multiplexed format for the selected SNPs and analysed ten maternal plasma samples from pregnancies at risk. The presence or absence of the paternal mutant allele was correctly determined in 27 out of 34 samples analysed. With haplotype analysis, NIPD was possible on eight out of ten families. This is the first study carried out for the NIPD of β-thalassaemia using targeted NGS and haplotype analysis. Preliminary results show that NGS is effective in detecting paternally inherited alleles in the maternal plasma.
Similar content being viewed by others


Introduction
β-Thalassaemia is one of the most common autosomal recessive single-gene disorders in Cyprus, where about 12% of the population are carriers.1 Currently, fetal genetic material for prenatal diagnosis is sampled by invasive procedures, which are associated with a significant risk of induced abortion.2, 3 The discovery that during pregnancy there is a median of 10% of cell-free fetal DNA in the maternal circulation opened up new avenues in the diagnostics of fetal genetics.4, 5, 6 However, this poses a technical challenge as fetal DNA represents a minor population in maternal plasma, exacerbated by the fragmentation of fetal DNA.7, 8
As the advent of next generation sequencing (NGS), a lot of effort has been concentrated on exploiting this technology for the measurement of aneuploidies in maternal plasma.9, 10, 11, 12, 13, 14 Using this technology, non-invasive prenatal diagnostic (NIPD) for trisomy 21, 18 and 13 has recently reached the clinical setting by analysing the relative amount of chromosomes in circulating cell-free DNA from maternal plasma.15, 16
However, approaches permitting reliable detection of single-gene mutations or single-nucleotide polymorphisms (SNPs) using cell-free fetal DNA in maternal plasma are still under development. This is considerably more difficult because it concerns fetal genetic changes that differ only slightly from the maternal genome. Recently, a number of different strategies have been investigated to meet the challenges for the non-invasive detection of β-thalassaemia using maternal plasma. Allele-specific real-time PCR is one of the first approaches that have been used to exclude paternal mutations in the maternal circulation.17 Preferential detection of fetal alleles was achieved through initial enrichment of fetal DNA,8, 18 while others enhanced the production of the mutated fetal allele by employing either peptide nucleic acid probes19 or COLD PCR.20 In the specific case of β-thalassaemia, MALDI-TOF mass spectrometry has been also investigated.8
Moreover, Lun et al21 employed digital size selection to investigate the relative mutation dosage for NIPD of β-thalassaemia. Our group employed the APEX/thalassochip approach, based on the detection of polymorphic SNPs, in order to successfully identify the paternally inherited allele of the fetus in the maternal plasma22, 23 while, more recently, Phylipsen et al24 employed pyrophosphorolysis activated polymerisation analysis using polymorphic SNPs to detect the paternal allele in maternal plasma of β-thalassaemia carriers. However, more SNPs need to be included in the study to link the paternal allele with minimal risk of misdiagnosis. Hence, despite the advances in technology, NIPD of β-thalassaemia has yet to reach clinical practice.8, 18, 19, 22, 23, 25
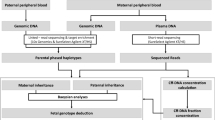
Our approach is based on the detection of the paternally inherited alleles as maternal alleles cannot be differentiated from fetal ones. Therefore, NIPD is possible only in those cases where the fetus inherits the normal allele of the father. The aim of this study is to assess the analytical power and specificity of a modified version of NGS using the Illumina platform, called ‘targeted sequencing’, for the reliable detection of paternally inherited SNPs in the maternal plasma of at risk pregnancies for β-thalassaemia. Moreover, this study aims to use this platform for the development of a fast and cost-effective non-invasive diagnostic assay for β-thalassaemia. The principle of our targeted sequencing is to selectively amplify or capture targeted regions of a DNA before sequencing. For this purpose, we have developed a method of integrating selectively amplified targeted DNA regions and NGS. The proof of principle results presented in this study show that the detection of paternally inherited SNPs in the maternal plasma is possible and reliable using targeted sequencing.
Materials and methods
Sample collection and processing
The study was approved by the Cyprus National Bioethics Committee and all subjects gave informed consent. Blood samples, as well as corresponding chorionic villi samples (CVS), were collected from ten families (including parents and grandparents) at risk for β-thalassaemia in their newborns. Approximately 9 ml maternal blood samples were collected into EDTA-containing tubes between the 10th and 11th week of gestation and before chorionic villus sampling. Plasma was separated from cells by centrifugation at low speed, 2500 g for 10 min without braking. It was transferred to microcentrifuge tubes and subjected to a second centrifugation step at 16 000 g for 40 min to remove any residual cells. The two centrifugation steps were performed within 4–8 h of collection.
DNA extraction
Cell-free DNA was extracted from 1 ml of maternal plasma using QIAamp Circulating Nucleic Acid Kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s instructions. Genomic DNA was extracted using the Puregene Blood Core Kit C (Qiagen Sciences, Germantown, MD, USA).
Selection of SNPs and primer design
Four SNPs, rs3834466_IIɛ, rs968857_3′ψβ, rs10768683_AvaII and rs7480526_II74, located on the β-globin gene cluster showing high heterozygosity in the Cypriot population were selected for analysis (Figure 1). SNPs rs968857_3′ψβ and rs7480526_II74 were selected after a genotyping analysis carried out in a previous study,26 whereas SNPs rs10768683_AvaII and rs3834466_IIɛ are routinely used in Cyprus for prenatal diagnosis.27 Two of the SNPs are located within the β-globin gene and the other two SNPs 5′ to the δ-globin gene, therefore, any recombination events would be instantly recognisable.

Four SNPs located on the β-globin gene cluster used in targeted NGS analysis.
Primer sequences were designed using the reference sequence of haemoglobin gene locus with accession number NG_000007 from the NCBI database (http://www.ncbi.nlm.nih.gov/nuccore/NG_000007). The largest PCR-fragment generated was no more than 268 bp (Supplementary Table 1).
PCR amplification
Targeted PCR was performed for the selected SNPs with the corresponding primers. Each reaction was carried out with 5 ng of genomic DNA or 1–5 ng of maternal plasma DNA in a total reaction volume of 25 μl that contained 1 × PCR buffer with 1.5 mM MgCl2, 200 μ M dNTPs and one unit of AmpliTaq Gold DNA polymerase (Applied Biosystems by Roche Molecule Systems Inc., Branchburg, NJ, USA). The amplification procedure consisted of an initial denaturation step at 95 °C for 10 min, followed by 45 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, extension at 72 °C for 30 s, followed by a final extension step at 72 °C for 7 min . The PCR products were purified using the MinElute PCR purification kit, Qiagen, according to the manufacturer’s instructions.
We used a ‘spiking’ approach with genomic DNA to determinate the specificity of the assay for each of the four SNPs. We used 100% genomic DNA having a homozygous genotype for the SNP and a sample of 90% homozygous DNA spiked with 10% DNA having a heterozygous genotype. For the confirmation of the reproducibility, four replicates were performed for each sample per SNP.
Nested PCR
New genomic primers for the nested PCR were designed using NCBI primer-Blast software so that the SNP of interest was within 36 bp from the nested primer. All genomic primers contained the sequence for the Illumina adaptors and the reverse primers also contained a 6 bp Illumina index barcode. For rs3834466_IIe, rs968857_3′ψβ and rs10768683_Ava the Illumina index barcodes 1, 2, 4, 5, 6, 7, 8, 9, 10 and 12 were used. For rs7480526_II74, the Illumina index barcodes 2, 4, 5, 6, 7, 8, 9, 12, 13 and 14 were used (Supplementary Table 1).
One microliter of the targeted PCR products was loaded on an Agilent Technologies 2100 Bioanalyzer (Santa Clara, CA, USA) using a DNA 1000 assay to determine the concentration and to check for quality. Nested PCR was performed to introduce the Illumina adaptors and the index barcode to the PCR products. Each reaction was carried out with 0.5 ng of targeted PCR products in a total reaction volume of 50 μl that contained 1 × phusion HF buffer, 200 μ M dNTPs and one unit Phusion DNA Polymerase (Finnzymes part of Thermo Fisher Scientific, Waltham, MA, USA). The nested PCR consisted of an initial denaturation step at 98 °C for 30 s followed by 18 cycles of denaturation at 98 °C for 10 s, annealing at 57.4 °C for 30 s, extension at 72 °C for 30 s, followed by a final extension step at 72 °C for 5 m. The PCR was performed in a Biometra TProfessional (Goettingen, Germany). The PCR fragments were purified using AMPure XP beads according to the manufacturer’s instructions. The products were eluted in 15 μl of elution buffer. One microliter of the product was loaded on an Agilent Technologies 2100 Bioanalyzer using a DNA 1000 assay to determine the quality and the quantity of the genomic library.
Bridge amplification and sequencing-by-synthesis
Cluster generation was performed according to the Illumina TruSeq SR Cluster kit v2 (cBot) Reagents Preparation Guide (www.illumina.com). Briefly, 40 PCR libraries were pooled together to get a stock of 10 nM. One microliter of the 10 nM stock was denaturated with NaOH, diluted to 6.5 pM and hybridised onto the flowcell. The hybridised products were sequentially amplified, linearised and end-blocked according to the Illumina Single Read Multiplex Sequencing user guide. After hybridisation of the sequencing primer, sequencing-by-synthesis was performed using the HiSeq 2000 with a 36-cycle protocol. The sequenced fragments were denaturated with NaOH using the HiSeq 2000 and the index-primer was hybridised onto the fragments. The index was sequenced with a 7-cycle protocol.
Sequence analysis
We used NARWHAL28 to demultiplex the raw data from the sequencer into FASTQ files per sample. For the spiked genomic samples, we did not perform any alignment, but we filtered the data to keep only reads in which all bases have at least a base-calling phred score of 10 and a mean phred score over the whole read of at least 30. We directly counted the number of times the different read sequences occurred. For the maternal plasma samples, we performed demultiplexing and BWA29 alignment against the human reference genome (version hg18) with NARWHAL and counted the occurrence of the reads for both alleles in the BWA aligned SAM files using standard Unix tools grep and sed. All subsequent calculations were performed in Microsoft Excel.
Results
Specificity of NGS on spiked samples
In order to develop a reliable method, we selected four SNPs in the β-globin locus, that are common in the Cypriot population, rs3834466_IIɛ, rs968857_3′ψβ, rs10768683_AvaII and rs7480526_II74, each showing high heterozygosity (Figure 1 and Materials and Methods). Next, we designed primers to specifically amplify the DNA containing these SNPs. Homozygous DNA was amplified directly or after spiking with heterozygous containing DNA, followed by Illumina sequencing and more than 1.5 million reads per sample were obtained (Table 1). In all homozygous samples for all SNPs (100%), the sequence reads match the expected homozygous sequence. In the spiked samples (90%+10%), the two different sequences present were accurately detected and differentiated in the expected (5%) allele concentration. We noted a somewhat higher concentrations for SNP rs7480526_II74 at around 8.2%. For this SNP, we also saw reads with an additional variant 7 bp before the SNP position. These variant reads have not been taken into account. A negligible variation was observed between replicates. We conclude that the analytical power of the platform is sufficiently high and specific to be tested on maternal plasma analysis.
Family study
The maternal and paternal genotypes for 101 families at risk were previously determined by MALDI-TOF MS for a panel of 49 SNPs.26 SNPs were considered informative when the mother was homozygous and the father was heterozygous for the same SNP. Ten families having at least two informative SNPs of the four analysed above were selected for the NGS analysis. SNPs where the mother was homozygous for one allele and the father was homozygous for the other allele were also included for confirmation of the paternal allele, whereas SNPs where both parents are homozygous were used for the determination of the sequencing error rate. SNPs where the mother was heterozygous were not used for the deduction of the paternal allele. The corresponding CVS material was also typed for the selected SNPs for the confirmation of the maternal plasma result.
Efficiency of NGS on maternal plasma
To detect the paternally inherited allele in the maternal plasma, we analysed the selected couples at risk of carrying a β-thalassaemia child for the corresponding informative SNPs. Each sample was analysed in triplicate. About two million reads were generated per sample replicate and the detailed results are shown in Table 2. The exact number of reads was determined for all 40 samples for both alleles (ten samples of four SNPs). The proportion of DNA molecules in the maternal plasma sample that originated from the fetus was determined based on the fractional fetal DNA (f) in the maternal plasma, which fetal is given by:

where p is the number of sequenced reads of the fetal specific allele and q is the read count of the other allele, which is shared by the maternal and fetal genomes.25 f was calculated from the sequencing data for each SNP and replicate (Table 2). Theoretically, a non-present fetal allele gives a calculated fetal fraction of 0, whereas the fetal fraction DNA in the maternal circulation can be as low as 3% with an average of 10%.6, 7 We used a cutoff of 2.5%, which is somewhat below the minimum value. We consider the fetal allele detected if f is larger than 2.5% for at least two out of three replicates.
All maternal genotypes determined from the sequence data were correct for the four SNP’s. Six sites were not used for the deduction of the paternal allele as the mother was heterozygous for those. The results for the fetal genotypes were compared with the results of the CVS analysis previously performed for prenatal diagnosis. From 34 samples analysed, concordance with CVS was observed in 27 cases, where we positively detected and differentiated the paternal allele in the maternal plasma in nine of the cases and in 18 cases no other measurable allele was observed (negative detection of the paternal allele as expected). However, we also observed four false-positive and three false-negative results. SNP rs3834466_IIɛ was analysed for nine samples as follows: for sample five the mother is heterozygous for the SNP and, therefore, the paternally inherited allele in the maternal background could not be discriminated. Six out of nine samples showed concordance with the CVS result, where we correctly detected the paternally inherited allele (true-positive detection) in three of the cases, whereas in three cases we correctly did not detect the paternal allele (true-negative detection). However, two false negatives and one false positive were observed. For SNP rs968857-3′ψβ, concordance with the CVS was observed in seven out of ten analysed samples with positive detection of the paternally inherited allele in five cases and negative detection in two cases. Two false positives and one false negative were observed. SNP rs10768683_AvaIIβ was analysed for eight samples showing correct negative detection of the paternally allele in seven cases, whereas in one case a single false positive detection was found, although this analysis appears suspect due to the very high positive score in one of the samples of the triplicate. For SNP rs7480526_II74, seven out of ten samples were analysed showing concordance with CVS in all samples analysed. We observed true-positive detection of the paternal allele in a single sample and true-negative detection in six of the cases.
Non-invasive fetal haplotyping
To investigate the feasibility of SNPs for the NIPD of β-thalassaemia analysed by Illumina sequencing of the maternal plasma, haplotype analysis was performed. The paternal haplotype was determined from previous family studies in our lab for prenatal diagnostic purposes. Based on the results obtained from NGS of maternal plasma, the haplotypes of the fetus were generated and the alleles of the fetus were correctly linked to the paternal normal or β-thal allele for eight out of ten families (Table 3). The haplotypes were inferred if two out of three or three out of four SNPs had the expected result and given that paternal alleles could be differentiated. More specifically, for families 3, 5, 8 and 10 the paternally inherited allele of the fetus was correctly linked based on the result of all SNPs analysed. For families 1, 6, 7 and 9, the fetal allele was correctly linked to the paternal, even though one of the SNPs analysed gave incorrect result. In these cases, the information obtained from the other SNPs was sufficient to link the fetal allele with the paternal one. However, for families 2 and 4, the fetal allele could not be linked as either more than one SNP showed an unexpected result (family 2) or there was insufficient information from the analysed SNPs (family 4). In these cases, the NIPD was inconclusive.
For families 1, 3, 6, 7 and 10, the fetal allele was correctly linked to the paternal β-thal allele and, consequently, the inheritance of the mutated paternal allele indicated that NIPD for these cases is β-thal trait or major. Therefore, direct invasive prenatal diagnosis in a fetal sample was recommended to confirm the diagnosis of the fetus. For families 5 and 9, the fetal allele is correctly linked to the normal allele of the father, indicating that NIPD is normal or β-thal trait and, therefore, invasive prenatal procedures may be avoided. We concluded that more than four SNPs and more replicates are needed to develop a reliable assay for haplotype analysis. The above analysis was only used as a model in order to demonstrate the effective linkage of the fetal allele to the paternal based on SNPs. In a diagnostic setting, where more SNPs and more replicates will be included per family, a diagnostic algorithm would have to be included in order to derive the final haplotypes and, in turn, the final diagnosis.
Discussion
Most NIPD studies carried out on β-thalassaemia were based on detection of the paternally inherited mutation and, therefore, limited to the couples sharing different mutations.17, 18, 19, 20 In view of this limitation, we previously showed that the detection of the paternally inherited SNPs is feasible for the NIPD of β-thalassaemia.22, 23 SNPs can be used regardless of the mutation of the carrier couples, they provide positive detection of the paternal allele, normal or mutant, the result can be confirmed with more than one SNP and, importantly, the more SNPs used the less diagnostic risk.
In this study, we took advantage of the analytical power of NGS Illumina platform to reliably detect and quantify all the sequences present in a sample to detect the paternally inherited SNPs in the maternal plasma of β-thal carriers.
The specificity of the platform to detect and differentiate the minor allele present in the overwhelming background of the maternal allele was confirmed using spiked genomic samples. This demonstrates the extreme precision and analytical power of the method, as the correct result in all samples was obtained with insignificant variation between the replicates.
The reliability of the method was assessed with a preliminary analysis of maternal plasma samples. The current study showed that the detection of paternally inherited allele in the maternal plasma is possible with the use of targeted sequencing and SNPs.
Haplotype analysis based on the sequence results showed diagnosis was possible for eight out of ten families. The importance of having a high number of SNPs for each family was illustrated in all cases. In four of the cases, the paternal inheritance of the fetus was correctly deduced based on all the SNPs analysed. However, in four cases where one SNP gave unexpected result, the paternally inherited allele was correctly linked based on the information given by the other three SNPs with an insignificant risk of misdiagnosis. In two cases where NIPD was inconclusive, the fetal allele could not be linked to the paternal one as more than one SNP gave a deviating result or there was lack of adequate information from the analysed SNPs. It is important to emphasise that no misdiagnosis was made even in the cases where some SNPs gave an incorrect result. Including a higher number of SNPs will further increase the statistical power to differentiate the maternal from the paternal allele through haplotype analysis with a higher level of accuracy and for a greater proportion of carrier couples. Such an increase can be easily incorporated in the current Illumina technology platform with only a small increase in costs.
However, further studies are needed in order to improve the reliability of the used assay to eliminate the false positives and negatives. Possible reasons for the observed false negatives are the inefficiency in the isolation of fetal material in the maternal plasma and the inefficiency of the DNA amplification due to its very small size and quantity. In some cases, the percentage of the minor paternal allele, as measured with the targeted sequencing approach, might be smaller than the actual percentage causing changes in the ratio of the alleles and, in turn, creating problems in their analysis. This depends on the position of the SNP on the amplicon in terms on where the fetal fragment is cut, as well as on the size of the fetal fragment. Fetal-derived DNA molecules are <300 bp7 showing a prominence at 143 bp.25 The amplicon sizes of our fragments were between 170 bp and 268 bp and therefore, fragments longer than 146 bp, in combination with the position of the SNP on the amplicon, are expected to result in the amplification of only a fraction of the fetal molecules present in the maternal plasma resulting in false-negative results.
Cross contamination between samples that might have arisen during the extraction process could be a possible cause of false positives, but highly unlikely here as it would have been observed in other SNPs for the same samples. False-positive results and erroneous base calls have been observed by other teams that have used different platforms for the analysis of cell-free fetal DNA but also the technology of NGS.9, 10, 15, 16 Palomaki et al16 and Chiu et al10 reported false-positive rates of 1.4% and 2.1%, respectively, which were improved in a subsequent study.9 False-positive SNP calls could be derived from erroneous alignments of short reads.30 Therefore, different bioinformatics parameters, as well as improvements, in statistical analysis have been investigated by these teams in order to eliminate the erroneous calls and decrease the observed false-positive rate. In order to accomplish that larger scale studies would need to be performed. Moreover, the PCR amplification used in this study might also be a cause of increased error rates by amplifying nonspecific fragments due to high number of cycles used, although the nested PCR partially eliminates unspecific amplified fragments from the first PCR reaction.
We have also observed some variations in the reads of sequenced fragments from sample to sample and from replicate to replicate. It is unclear at this point whether this stems from the quality of the sample, PCR artefacts because of the high number of amplification cycles or during sequencing library preparation or cluster generation.
To avoid the observed discrepancies and to improve the diagnostic efficiency, it is suggested to include a higher number of maternal samples, as well as more SNPs and more replicates per sample with improved conditions for each SNP. This will also aid to derive statistical cutoff values specific for the data of each SNP as opposed to fixed cutoff values. Furthermore, it is suggested to have fragments around 143 bp in size in order to capture and amplify the maximum of the fetal molecules present in the maternal plasma. Furthermore, it is recommended to use less PCR cycles in order to avoid the introduction of erroneous bases. Finally, in future experiments free DNA isolated from plasma of non-pregnant women negative for the SNPs can also be included for a better evaluation of the false positives.
In this study, we have outlined and demonstrated the analytical power of NGS and targeted sequencing for the analysis of fetal DNA sequences in the maternal plasma. The accuracy and precision demonstrated allowed us to detect and differentiate the paternally inherited allele of the fetus in the overwhelming background of the maternal alleles. The linkage of paternally inherited allele was possible based on haplotype analysis, provided that other family members or a previously born child were available for testing. The group of Lam31 employed the relative haplotype approach to deduce the paternally inherited allele without the need of other family members. However, their procedure is complicated and costly. Although the NGS method is a complex method, it has been demonstrated that it can be used to detect maternal mutations with accuracy and precision.25, 31, 32, 33 Moreover, the targeted sequencing protocol using short 36 bp reads is a faster and more cost-effective than whole genome amplification used by Lo et al25 for NIPD of β-thalassaemia. However, in order to implement our targeted SNP sequencing assay in diagnostics the method has to be simplified. The continuous announcements of smaller, more personalised NGS platforms promise new low-cost, rapid and less complicated sequencing accessible to more laboratory settings.
Directions for future development would be to improve the diagnostic efficiency, precision, accuracy and reliability by eliminating false positives and false-negative results. Finally, as this approach applies only to the 50% of the cases where the fetus inherits the normal allele of the father, in the future one should look for other approaches using this technology to also deduce the maternally inherited allele.
References
Kyrri EK, Loizidou D, Ioannou C, Makariou C, Kythreotis LM, Phylactides PK et al: The changing epidemiology of β-thalassaemia in the Greek-Cypriot population. Hemoglobin 2013, (in press).
Alfirevic Z, Sundberg K, Brigham S : Amniocentesis and chorionic villus sampling for prenatal diagnosis. Cochrane Database Syst Rev 2003; CD003252.
Tabor A, Philip J, Madsen M, Bang J, Obel EB, Norgaard-Pedersen B : Randomised controlled trial of genetic amniocentesis in 4606 low-risk women. Lancet 1986; 1: 1287–1293.
Lo YM, Corbetta N, Chamberlain PF et al: Presence of fetal DNA in maternal plasma and serum. Lancet 1997; 350: 485–487.
Lun FM, Chiu RW, Allen Chan KC, Yeung Leung T, Kin Lau T, Dennis LoYM : Microfluidics digital PCR reveals a higher than expected fraction of fetal DNA in maternal plasma. Clin Chem 2008; 54: 1664–1672.
Lo YM, Tein MS, Lau TK et al: Quantitative analysis of fetal DNA in maternal plasma and serum: implications for noninvasive prenatal diagnosis. Am J Hum Genet 1998; 62: 768–775.
Chan KC, Zhang J, Hui AB et al: Size distributions of maternal and fetal DNA in maternal plasma. Clin Chem 2004; 50: 88–92.
Li Y, Di Naro E, Vitucci A et al: Size fractionation of cell-free DNA in maternal plasma improves the detection of a paternally inherited beta-thalassemia point mutation by MALDI-TOF mass spectrometry. Fetal Diagn Ther 2009; 25: 246–249.
Chen EZ, Chiu RW, Sun H et al: Noninvasive prenatal diagnosis of fetal trisomy 18 and trisomy 13 by maternal plasma DNA sequencing. PLoS One 2011; 6: e21791.
Chiu RW, Akolekar R, Zheng YW et al: Non-invasive prenatal assessment of trisomy 21 by multiplexed maternal plasma DNA sequencing: large scale validity study. BMJ 2011; 342: c7401.
Chiu RW, Chan KC, Gao Y et al: Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc Natl Acad Sci USA 2008; 105: 20458–20463.
Fan HC, Blumenfeld YJ, Chitkara U, Hudgins L, Quake SR : Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc Natl Acad Sci USA 2008; 105: 16266–16271.
Fan HC, Quake SR : Sensitivity of noninvasive prenatal detection of fetal aneuploidy from maternal plasma using shotgun sequencing is limited only by counting statistics. PLoS One 2010; 5: e10439.
Chiu RW, Sun H, Akolekar R et al: Maternal plasma DNA analysis with massively parallel sequencing by ligation for noninvasive prenatal diagnosis of trisomy 21. Clinical Chem 2010; 56: 459–463.
Palomaki GE, Deciu C, Kloza EM et al: DNA sequencing of maternal plasma reliably identifies trisomy 18 and trisomy 13 as well as Down syndrome: an international collaborative study. Genet Med 2012; 14: 296–305.
Palomaki GE, Kloza EM, Lambert-Messerlian GM et al: DNA sequencing of maternal plasma to detect Down syndrome: an international clinical validation study. Genet Med 2011; 13: 913–920.
Chiu RW, Lau TK, Leung TN, Chow KC, Chui DH, Lo YM : Prenatal exclusion of beta thalassaemia major by examination of maternal plasma. Lancet 2002; 360: 998–1000.
Li Y, Di Naro E, Vitucci A, Zimmermann B, Holzgreve W, Hahn S : Detection of paternally inherited fetal point mutations for beta-thalassemia using size-fractionated cell-free DNA in maternal plasma. JAMA 2005; 293: 843–849.
Galbiati S, Foglieni B, Travi M et al: Peptide-nucleic acid-mediated enriched polymerase chain reaction as a key point for non-invasive prenatal diagnosis of beta-thalassemia. Haematologica 2008; 93: 610–614.
Galbiati S, Brisci A, Lalatta F et al: Full COLD-PCR protocol for noninvasive prenatal diagnosis of genetic diseases. Clin Chem 2011; 57: 136–138.
Lun FM, Tsui NB, Chan KC et al: Noninvasive prenatal diagnosis of monogenic diseases by digital size selection and relative mutation dosage on DNA in maternal plasma. Proc Natl Acad Sci USA 2008; 105: 19920–19925.
Papasavva T, Kalakoutis G, Kalikas I et al: Noninvasive prenatal diagnostic assay for the detection of beta-thalassemia. Ann N Y Acad Sci 2006; 1075: 148–153.
Papasavva T, Kalikas I, Kyrri A, Kleanthous M : Arrayed primer extension for the noninvasive prenatal diagnosis of beta-thalassemia based on detection of single nucleotide polymorphisms. Ann N Y Acad Sci 2008; 1137: 302–308.
Phylipsen M, Yamsri S, Treffers EE et al: Non-invasive prenatal diagnosis of beta-thalassemia and sickle-cell disease using pyrophosphorolysis-activated polymerization and melting curve analysis. Prenatal Diagnosis 2012; 32: 578–587.
Lo YM, Chan KC, Sun H et al: Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med 2010; 2: 61ra91.
Papasavva TE, Lederer CW, Traeger-Synodinos J et al: A minimal set of SNPs for the noninvasive prenatal diagnosis of beta-thalassaemia. Ann Hum Genet 2013; 77: 115–124.
Old JM, Petrou M, Modell B, Weatherall DJ : Feasibility of antenatal diagnosis of beta thalassaemia by DNA polymorphisms in Asian Indian and Cypriot populations. Br J Haematol 1984; 57: 255–263.
Brouwer RW, van den Hout MC, Grosveld FG, van Ijcken WF : NARWHAL, a primary analysis pipeline for NGS data. Bioinformatics 2012; 28: 284–285.
Li H, Durbin R : Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760.
Dohm JC, Lottaz C, Borodina T, Himmelbauer H : Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acid Res 2008; 36: e105.
Lam KW, Jiang P, Liao GJ et al: Noninvasive prenatal diagnosis of monogenic diseases by targeted massively parallel sequencing of maternal plasma: application to beta thalassemia. Clin Chem 2012; 58: 1467–1475.
Fan HC, Gu W, Wang J, Blumenfeld YJ, El-Sayed YY, Quake SR : Non-invasive prenatal measurement of the fetal genome. Nature 2012; 487: 320–324.
Kitzman JO, Snyder MW, Ventura M et al: Noninvasive whole-genome sequencing of a human fetus. Sci Transl Med 2012; 4: 137ra176.
Acknowledgements
We thank Mrs Elena Kyriacou for her secretarial assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Papasavva, T., van IJcken, W., Kockx, C. et al. Next generation sequencing of SNPs for non-invasive prenatal diagnosis: challenges and feasibility as illustrated by an application to β-thalassaemia. Eur J Hum Genet 21, 1403–1410 (2013). https://doi.org/10.1038/ejhg.2013.47
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.47
Keywords
This article is cited by
-
Noninvasive prenatal diagnosis targeting fetal nucleated red blood cells
Journal of Nanobiotechnology (2022)
-
Identification of a de novo fetal variant in osteogenesis imperfecta by targeted sequencing-based noninvasive prenatal testing
Journal of Human Genetics (2018)
-
Noninvasive prenatal diagnosis of 21-Hydroxylase deficiency using target capture sequencing of maternal plasma DNA
Scientific Reports (2017)
-
Non-invasive prenatal diagnosis of beta-thalassemia by semiconductor sequencing: a feasibility study in the sardinian population
European Journal of Human Genetics (2017)
-
Non-invasive prenatal diagnosis of β-thalassemia by detection of the cell-free fetal DNA in maternal circulation: a systematic review and meta-analysis
Annals of Hematology (2016)
