
Abstract
Mendez and colleagues reported the identification of a Y chromosome haplotype (the A00 lineage) that lies at the basal position of the Y chromosome phylogenetic tree. Incorporating this haplotype, the authors estimated the time to the most recent common ancestor (TMRCA) for the Y tree to be 338 000 years ago (95% CI=237 000–581 000). Such an extraordinarily early estimate contradicts all previous estimates in the literature and is over a 100 000 years older than the earliest fossils of anatomically modern humans. This estimate raises two astonishing possibilities, either the novel Y chromosome was inherited after ancestral humans interbred with another species, or anatomically modern Homo sapiens emerged earlier than previously estimated and quickly became subdivided into genetically differentiated subpopulations. We demonstrate that the TMRCA estimate was reached through inadequate statistical and analytical methods, each of which contributed to its inflation. We show that the authors ignored previously inferred Y-specific rates of substitution, incorrectly derived the Y-specific substitution rate from autosomal mutation rates, and compared unequal lengths of the novel Y chromosome with the previously recognized basal lineage. Our analysis indicates that the A00 lineage was derived from all the other lineages 208 300 (95% CI=163 900–260 200) years ago.
Similar content being viewed by others

Main
‘Look, if I told you that I keep a goat in the backyard of my house… and if you happened to have a man nearby, you might ask him to look over my garden fence… But what would you do if I said, ‘I keep a unicorn in my backyard’?’
James Randi (Maddox, Randi, and Stewart 1988)
Introduction
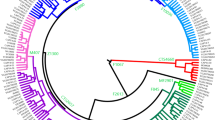
The Y chromosome of a descendent of Albert Perry, an African American from South Carolina (born circa 1819–1827),1 was recently identified as representing an out-group lineage to all other known Y haplotypes presently identified in the human population.2 We will refer to the Y chromosome as Perry’s Y chromosome because the region of the Y that was examined is the X-degenerate, non-recombining portion of the Y, expected to be nearly identical between Albert Perry and his male descendants. This Y haplotype was dubbed A00, in reference to a previously recognized oldest lineage that was rebranded as A0. The identification of a novel Y haplotype is always exciting, and this new haplotype, in particular, is unique in its basal position on the Y haplotype tree, which justifies its moniker ‘the Y-chromosomal Adam haplotype’. However, the announcement by Mendez et al2 that the coalescent time of all human Y chromosomes or the time to the most recent common ancestor (TMRCA) is approximately 338 000 years ago (ya), with a 95% confidence interval of 237 000–581 000 ya, was surprising on many levels. First, this estimate is more than double the oldest previous estimate of 141 500±15 600 ya,3 and is hugely larger than all the other previous or subsequent estimates, which ranged from 46 000 to 160 000 ya.4, 5, 6, 7, 8 Second, it significantly predated the most ancient mitochondrial DNA, which Poznik et al7 had recently estimated to be only slightly younger than the Y chromosome (mtDNA:99 000–148 000 vs Y:120 000–156 000 ya). Third, this TMRCA estimate is 142 000 years older than the oldest known anatomically modern human, estimated to be 196 000±2000 years old.9 Thus, this TMRCA inference suggests that either this Y chromosome is from a different ‘species’ (sensu Hammer),10 or that the ancestral population of anatomically modern Homo sapiens became subdivided into genetically differentiated subpopulations much earlier than previously known.2 One of the authors of Mendez et al2 even proposed that early Homo sapiens mated with ‘an unknown archaic species in western Central Africa’.10 Although either of the two scenarios above may be true,11, 12 there is no scientific support for either one for the Y chromosome. We wondered whether a simpler explanation might exist.
Here, we reassess the data and methodology in Mendez et al.2 In particular, we discuss (1) the decision to derive the Y-specific substitution rate from autosomal mutation rates instead of using previously inferred Y-specific substitution rates; and (2) the use of sequences of unequal lengths in the comparison between A00 and the previously recognized basal lineage, A0.
We uncover several methodological irregularities and analytical biases, each of which, have inflated the TMRCA estimate. By correcting these, we infer the new Y lineage characterized by Mendez et al2 to be significantly younger than originally reported.
Results and Discussion
In the following, we outline the various assumptions made in Mendez et al2 and their effect on estimating Y TMRCA.
(1) The decision to derive the Y-specific substitution rate from autosomal mutation rates instead of using previously inferred Y-specific substitution rates
Computing divergence time estimates is not different, in principle, from computing the time it takes two cars traveling in opposite directions at known speeds to reach a certain distance from each other. The time inferences will be overestimated if the distance between the two cars is overestimated, or if the speed of either car is underestimated. Similarly, biological divergence times will seem larger than the actual divergence times if genetic distances between sequences are overestimated or if the rates of substitution are underestimated.
In their study, Mendez et al2 could have used existing estimates for Y-specific substitution rates in the literature.4, 13, 14 Instead, they derived a substitution rate for the Y chromosome (6.12 × 10−10) using autosomal mutation rates reported from an Icelandic data set of parent-offspring trios in which one child is either autistic or schizophrenic.15 Interestingly, the authors even acknowledge that the TMRCA would have been much shorter had they used the Y-specific mutation rate in the literature. For example, Xue et al13 sequenced approximately 10.15 Mb from two Y chromosomes of two European individuals separated by 13 generations and inferred a substitution rate of 1 × 10−9 substitutions per nucleotide per year, under the assumption that the generation time is 30 years. This estimate is consistent with estimates derived from human-chimpanzee Y chromosome analyses (1.5 × 10−9−2.1 × 10−9 substitutions per nucleotide per year) under the assumption of a divergence time range of 5–7 million years.16, 17
A substitution rate estimate of 1 × 10−9 substitutions per nucleotide per year is a widely accepted estimate. For example, Wei et al18 applied this rate to estimate that the time depth of the Y-chromosomal tree was ∼101 000–115 000 years, and dated the lineages found outside Africa to 57 000–74 000 years. Cruciani et al3 also applied this substitution rate to derive an estimate of ∼142 000 years to the TMRCA of the Y chromosome.
Substitution rate on the Y chromosome is not linearly related to the autosomal rate
Evolutionarily, as the autosomes and the sex chromosomes spend different amounts of time in the paternal and maternal lineages, male mutation bias can be approximated by comparing the substitution rates on any two pairs of these chromosome types. If differences in replication alone can explain all of the differences between the substitution rates on the sex chromosomes and autosomes, then the magnitude of male mutation bias should be the same regardless of which chromosomes are compared. Although male mutation bias may explain most of the differences in the substitution rates between each sex chromosome and the autosomes, it cannot account for all of the rate variation; the magnitude of male mutation bias will vary significantly depending on which pair of chromosome types are compared.19, 20, 21 If estimates of male mutation bias vary depending on which chromosome types are compared, then factors other than replication must affect mutation rates on each chromosome type, and one cannot assume a direct correlation between the mutation rates of any chromosome types, as did Mendez et al.2
The assumption that mutation rates are equal to substitution rates
Mutation rate refers to the rate at which changes in the nucleotides are incorporated into the DNA sequence during replication, that is, the probability that an allele differs from the copy of that in its parent from which it was derived. Substitution rate refers to the rate at which a newly arisen allele is incorporated into a population, for example, when a newly arisen allele becomes fixed in a population. This rate is equal to experimentally measured apparent mutation rates only for fairly short times when recurring mutations, purifying selection, and genetic drift are negligible. Otherwise, those effects should be considered and corrected.
Using the single-generation mutation rates calculated by Kong et al15, Mendez et al,2 developed a likelihood-based method, which was used to estimate the TMRCA of A00 and that of the common ancestor of African Americans and individuals belonging to the Mbo ethnic group of Cameroon. For these analyses, Mendez et al2 presumed that long-term substitution rates are equal to single-generation mutation rates as determined by Kong et al,15 and assumed a complete lack of purifying or advantageous selection on the Y chromosome. Is this a reasonable assumption?
Substitution rates vary among chromosome types depending on several factors. A principal contributor to substitution rate is the mutation rate, which in turn is determined by the number of germ-line replications between successive generations. An additional determinant of substitution rate is the efficacy of purifying selection, which in turn depends not only on the particular constraints of each chromosome, but also on the long-term effective population size for each chromosome type. For example, the long-term effective population sizes for the X and Y chromosomes are, respectively, three-quarter and one-quarter of the long-term effective population size of autosomes.
Purifying selection is expected to remove deleterious mutations as well as linked neutral variation,22 which may have confounding effects in the Y chromosome. Unlike the autosomes, most of the Y chromosome is non-recombining, so purifying selection is less efficient there.23, 24 Moreover, the repetitive structure of the Y chromosome, where gene conversion may occur between repetitive palindrome arms,17 makes deletions events highly frequent.25 For example, recombination between homologous sequences in palindromes on the Y chromosome frequently removes 6–7 Mb of sequence and several fertility genes.26 The inefficiency of purifying selection coupled with frequent, recurrent mutations has likely allowed many deleterious mutations to reach high frequency in humans.25, 27 However, most of the Y chromosome is non-recombining; thus, the effects of purifying selection acting anywhere on the Y chromosome will be magnified, because all linked neutral variation will also be removed.22 As such, the actual diversity may be lower than expected given the Y-specific mutation rate.28
Genetic drift is another powerful force that shapes the genetic diversity of the Y chromosome and haplotype groups and, in turn, depends on stochastic dynamics and social selection.29, 30 Arising through stochastic variation in the number of offspring, the effect of genetic drift is much stronger for the Y chromosome than for autosomal segments, because only one copy may be passed on to the next generation compared with four autosomal copies. Therefore, the opportunity for a stochastic change differs correspondingly and has profound effects on the Y chromosome, particularly when occurring in small populations with low effective deme sizes, such as those that have characterized humankind for most of its evolution.31 In the Mbo samples described by Mendez et al,2 as having A00 haplotypes, the main contribution to the variation was by genetic drift. Consequently, these haplotypes are derived from a common ancestor who lived only a few centuries ago (Mendez et al2), not 338 000 ya, when a common ancestor of A00 population was reported to live. In other words, as genetic drift causes a major impact on the apparent history of a population compared with that of mutation rates, it is incorrect to employ mutation rates obtained at relatively small time spans for long-term evolution process without considering other effects that might be important.
All of this means that, although the Y chromosome is expected to have a higher mutation rate due to more rounds of cell division and its existence in haploid non-recombining state, selective pressures and genetic drift might make it difficult to correctly infer its substitution rate from secondary data, such as autosomal substitution rates. Assuming a correlation between the mutation rate on the autosomes and on the Y chromosome, even when correcting for paternal age at conception, it may result in an underestimate of the Y-specific mutation rate.
Because assumptions about a correlation between autosomal- and Y-specific substitution rates are inconsistent with observations,19, 20 and because the substitution rate may be more reliably estimated from pedigree information,32 it would be ideal to measure the substitution rate for the Y chromosomes directly, as was done by Xue et al.13
The use of unreasonable generation times
To obtain a mean substitution rate for the Y chromosome per generation (μy) of 6.12 × 10−10 with a range of 4.39 × 10−10 ≤ μy≤7.07 × 10−10, Mendez et al2 assumed that anatomically modern human (AMH) males had a paternal generation time that, on average, ranged from 20 to 40 years. This assumption is extremely important (and problematic) since it affects estimates of the male mutation bias.
Male mutation bias (also referred to as ‘male driven evolution’) alludes to the higher rate of mutations in the male lineage versus the female lineage, resulting from the higher number of rounds of replication of the sperm relative to the rounds of replication of the eggs.33 Male mutation bias has been observed in all mammals studied to date and its magnitude was shown to increase with increasing generation time.20 In humans, the relevance of male mutation bias is particularly manifested in older fathers, whose offspring harbor more autosomal mutations than the offspring of younger fathers.15, 34 However, it is not clear whether the huge variation in paternal age at conception assumed by Mendez et al2 is a reasonable assumption in modern human populations, let alone in ancient ones. For instance, even among developed nations, where age at conception is delayed, generation times ranges from 20 to 30 years35 and stands at ∼25 in the US.36 Less developed nations exhibit much shorter generation times (in the low 20 s).35 For the vast majority of human history and until the modern era, women married anytime from their mid- to late-teens and likely had their first child by the age of 20.37 Ancient societies were almost as age demanding for males. The Augustan marriage laws, for example, penalized males who did not sire a child by the age of 25.38 It thus seems unlikely that the average age of ancestral human fathers was older than, or even equal to, modern humans, particularly due to the fact that the mean life expectancy of Cameroon males (37.2 years) was lower than the purported upper bound of the generation time.39 By using a lower bound of 20 years, an average of 30 years, and an upper bound of 40 years, Mendez et al2 reduced the number of generations per unit time, and further inflated the TMRCA estimate.
The use of confidence intervals rather than prediction intervals, and the use of 90% confidence interval rather than the customary 95% or 99%
Mendez et al2 based their estimates of Y-specific substitution rates on the autosomal mutation rates from Kong et al.15 In their conversion of one estimate to the other, they used a simple model, according to which the mutation rate in the female lineage is constant, while the mutation rate in the male lineage increased with age of the father. The Kong et al15 data contain five data points from which to compute the maternal rate of mutations per generation. In the five trios, the number of maternal mutations was 9, 10, 11, 15, and 26. From these five data points, Mendez et al2 calculated a ‘median’ rate of 14.2 and a ‘standard deviation’ of 3.12. However, the correct values are 11 for the median and 6.98 for the standard deviation.
In statistics, a confidence interval is an observed interval used to indicate the reliability of an estimate of interest, not its distribution. By contrast, a prediction interval is an estimate of an interval within which future observations will fall, with a certain probability, given what has already been observed. Consequently, prediction interval is always wider than the corresponding confidence interval because of the added uncertainty involved in predicting a single response versus the mean response. In other words, a confidence interval indicates that we have a certain confidence to find the population mean within a range, whereas prediction interval predicts with a certain confidence that the next sample would be included within a range. Given that the calculations of Mendez et al2 involved simulations and sampling, they should have used the prediction interval. Instead, Mendez et al2 computed an observed confidence interval, which is much narrower than the prediction interval, hence, making the result appear much more tightly clustered around the mean than they really are. In addition, Mendez et al2 used a 90% confidence interval, rather than the customary 95 or 99% intervals, thus artificially decreasing the dispersal around the mean. Finally, they assumed that the five observed data points from Kong et al15 are normally distributed, but it is hard to believe that normality can be deduced from five data points. Mendez et al2 did not provide any justification for their assumption of normality.
By using a 90% sample confidence interval, Mendez et al2 inferred that the number of mutations per generation in the female lineage ranges from 9.07 to 19.33. Had they used a 95 or a 99% interval, the ranges would have been 8.08–20.32 and 6.16–22.24, respectively. Had they used the correct prediction confidence interval with 95 or 99% confidence, the number of mutations per generation in the female lineage would have been 0.52 to 27.88 and −3.78 to 32.18, respectively, where the sign − denotes ‘minus’. The perplexing range −3.78 to 32.18 is due to the assumption that the number of mutations per generation follows a normal distribution. That is, the normality assumption of Mendez et al2 results in the time-bending possibility that the most common ancestor of all the Y chromosomes in the world has yet to be born.
Further, in the calculation of the substitution rate for the Y chromosome, Mendez et al2 used estimates of the number of maternal substitutions estimated from the maternal lineage from five families, but took the mean number of mutations from all 78 trios (63.2) instead of using the same five pedigrees (69.6), which results in fewer expected mutations from the paternal lineage, again decreasing the estimate of the mutation rate for the Y chromosome and inflating the TMRCA estimate.
Finally, if the authors truly wanted to consider the male mutation rate per year, from autosomal pedigree data, it would have been reasonable to compute it directly from the five pedigrees where mutations are partitioned into those of maternal and paternal origin. From these five pedigrees, dividing the number of paternally-derived de novo autosomal mutations by the total number of sites assayed by Kong et al,15 by the father’s age at conception, one can estimate paternal mutation rates that range from 5.57 × 10−10 to 8.65 × 10−10 mutations per site per year (Supplementary Table S1). Three of these five direct estimates are higher than the upper bound for the paternal mutation rate suggested by Mendez et al.2 Across all five mutation rates, one can obtain estimates for the Y chromosome TMRCA, including the newly identified lineage between 242 200 (194 200–297 500) and 376 200 (301 600–462 100) ya. Curiously, there is also quite a large variation in the maternal mutation rate, with estimates ranging from 1.46 × 10−10 to 3.07 × 10−10 mutations per site per year (Supplementary Table S1), suggesting a considerable amount of variation in the number of mutations observed in a single generation.
(2) The use of sequences of unequal lengths in the comparison between A00 and the previously recognized basal lineage, A0
Mendez et al2 sequenced a portion of Perry’s Y chromosome (A00) as well as the closest phylogenetic Y haplotype to identify private and derived mutations in this lineage. Interestingly, the authors counted mutations in ∼240 kb of the X-degenerate portion of the A00 chromosome, but only reported mutations for ∼180 kb of the A0 chromosome. We first note that a reliable evolutionary estimate cannot be obtained from 2% of the male-specific portion of the Y chromosome. Second, it is also unclear why the sequence of the previously known basal lineage is 25% shorter than the novel Y chromosome, given the author’s obvious intent to compare the two chromosomes. In fact, the A0 chromosome was originally sequenced to the full extent of the A00 chromosome, but the authors chose to omit 60 000 bases of it because they consist of ‘a large amount of mutations’ (FLM personal communication). Remarkably, they reported the mutations in the regions on the A00 chromosome for which the matching A0 regions were dropped. In Figure 1 and Supplementary Table S1 of Mendez et al,2 43 mutations were reported as derived for the A00 chromosome and 45 were A0 derived mutations. These mutations were divided into two types: A0T (18 mutations) in which A00 is the only ancestral lineage and A0 (27 mutations) mutations that are observed only in chromosomes that are in the A0 haplogroup, though some of these mutations may be absent from some A0 Y chromosomes. We believe that matching chromosomal regions should be compared instead of eliminating particular regions in an attempt to make the data fit a preconceived model. We further speculate that omitting regions for one lineage, but including them for another may have reduced their estimated age. Indeed, in our calculations below, we show that the TMRCA calculation using equivalent regions of A0 and A00 yields a much lower estimate than that reported by Mendez et al.2
Y chromosome TMRCA
Calculating TMRCA based on sequence data
Using an MLE model40 and given mutation counts of 45 (27 A0+18 A0T) and 43 (A00) and the pedigree-based Y-chromosomal mutation rate (1 × 10−9),13 we calculated the TMRCA for the Y lineage including the A00 chromosome as 209 500 (95% CI=168 000–257 400) ya. However, the mutation counts were obtained for uneven lengths of sequence. Looking only at the sites that overlap between A0 and A00, there are 44 (26 A0+18 A0T) and 31 mutations (A00), corresponding to a slightly more recent TMRCA of 208 300 (95% CI=163 900–260 200) ya. Interestingly, repeating the last calculation with the Y-chromosomal mutation rate of 1.33 × 10−9 proposed by Wilder, Mobasher and Hammer5 yields an even more recent TMRCA of 156 600 (95% CI=123 200–195 600) ya. Unfortunately, because we do not know the number of mutations omitted by the authors from the A0 lineage, we can only postulate that the actual age of the A00 lineage is within these estimates.
Fitting of TMRCA estimates of the Y with X, autosomal and mtDNA TMRCAs
It is not unreasonable to find regions of the genome with TMRCA estimates that exceed divergence from the fossil record. However, in this particular case, there is reason for additional consideration because not only is the TMRCA reported for human Y chromosomes by Mendez et al2 significantly older than the mtDNA chromosome and the fossil age of anatomically modern humans, it is also inconsistent with population genetic theory. In the following, we examine the fitting between the Y-chromosomal TMRCA calculated based on two mutation rates to that of other genomic regions (Table 1). Under assumptions of neutrality, the effective population size of the Y chromosome is expected to be equal to the effective population size of the mtDNA—one-quarter that of the autosomes and one-third that of the X chromosome. Current observations of the TMRCA across other genomic regions (Table 1) are incompatible with the high Y chromosome TMRCA computed using the derived Y chromosome mutation rate,2 but are consistent with a Y chromosome TMRCA calculated using the mutation rate estimated from a Y-pedigree.13 Our findings show that an estimate of TMRCA based on the pedigree-based Y-chromosomal mutation rate (1 × 10−9 mutations/nucleotide/year) is more consistent with TMRCA estimates calculated for other chromosome types. In addition, recent work has shown that the observed diversity on the entire Y chromosome is approximately one-tenth of the expected, due to the effects of selection acting to reduce diversity on this non-recombining chromosome.28 If selection is acting to reduce diversity on the Y, then the TMRCA estimates of Mendez et al2 are likely substantial underestimates, putting them even more at odds with estimates of the TMRCA on the mtDNA, X and autosomes.
Conclusions
Paleontological descriptions largely differ from the iconic gorilla-to-human linear evolution and even from a human family tree mode. In reality, the human phylogenetic tree contains a large gap between chimpanzee and Ardipithecus ramidus (4.3–4.4 million (m) ya) and smaller gaps in the nearest human tree, making it difficult to infer potential interactions. Nonetheless, it is clear that in the past million years, several lineages including perhaps the Homo erectus (‘Java man’) (0.6–0.2 mya), Homo heidelbergensis (‘Heidelberg Man’) (735–230 kya),41 Homo rhodesiensis (400–110 kya),42 and Homo neanderthalensis (‘Neanderthal’) (400–30 kya)43, 44 coexisted and interbred with each other leading to the appearance of the first AMH. In the Middle Paleolithic (∼100–200 kya), AMH like the Omo (195±5 kya)9 and the Homo sapiens idaltu (160–154 kya)45, 46 evolved from these archaic Homo sapiens and persisted alongside modern humans.47, 48 The question of whether and to what extent AMH interbred with their archaic predecessors is one of the most fascinating questions in anthropology.
We have shown that consistently throughout their examination, Mendez et al2 have chosen the assumptions, approximations, numerical miscalculations and data manipulation that inflated the final TMRCA estimate. We agree that Mendez et al,2 in collaboration with members of the public and the FamilyTreeDNA company, have identified a novel Y haplotype that pushes back the estimate of the Y-specific TMRCA further than previous studies. However, we argue that the autosomally-derived Y substitution rate lacks support, and show that the TMRCA estimate from sequence data should be 208 300 (95% CI=163 900–260 200 ya), which is within the time frame of the emergence of AMH, excluding the possibility of introgression with more ancient hominin taxa.
We too share the excitement that increased participation by people of all ethnicities in population genetic studies will yield additional discoveries of who we are and where we came from. We have, however, shown that when assessing new data, care must be taken in both data analysis and methodology to ensure that the results are scientifically robust.
References
FamilyTreeDNA: Y-Haplogroup A Project - Y-DNA Classic Charthttp://www.familytreedna.com/public/Haplogroup_A/default.aspx?section=yresults Vol 2013.
Mendez FL, Krahn T, Schrack B et al: An african american paternal lineage adds an extremely ancient root to the human y chromosome phylogenetic tree. Am J Hum Genet 2013; 92: 454–459.
Cruciani F, Trombetta B, Massaia A, Destro-Bisol G, Sellitto D, Scozzari R : A revised root for the human Y chromosomal phylogenetic tree: the origin of patrilineal diversity in Africa. Am J Hum Genet 2011; 88: 814–818.
Thomson R, Pritchard JK, Shen P, Oefner PJ, Feldman MW : Recent common ancestry of human Y chromosomes: evidence from DNA sequence data. Proc Natl Acad Sci USA 2000; 97: 7360–7365.
Wilder JA, Mobasher Z, Hammer MF : Genetic evidence for unequal effective population sizes of human females and males. Mol Biol Evol 2004; 21: 2047–2057.
Tang H, Siegmund DO, Shen P, Oefner PJ, Feldman MW : Frequentist estimation of coalescence times from nucleotide sequence data using a tree-based partition. Genetics 2002; 161: 447–459.
Poznik GD, Henn BM, Yee MC et al: Sequencing Y chromosomes resolves discrepancy in time to common ancestor of males versus females. Science 2013; 341: 562–565.
Klyosov AA, Rozhanskii IL : Re-examining the ‘out of africa’ theory and the origin of Europeoids (Caucasoids) in light of DNA genealogy. Adv Anthropol 2012; 2: 80–86.
McDougall I, Brown FH, Fleagle JG : Stratigraphic placement and age of modern humans from Kibish, Ethiopia. Nature 2005; 433: 733–736.
Hammer MF : Human hybrids. Sci Am 2013; 308: 66–71.
Yang MA, Malaspinas AS, Durand EY, Slatkin M : Ancient structure in Africa unlikely to explain Neanderthal and non-African genetic similarity. Mol Biol Evol 2012; 29: 2987–2995.
Green RE, Krause J, Briggs AW et al: A draft sequence of the Neanderthal genome. Science 2010; 328: 710–722.
Xue Y, Wang Q, Long Q et al: Human Y chromosome base-substitution mutation rate measured by direct sequencing in a deep-rooting pedigree. Curr Biol 2009; 19: 1453–1457.
Francalacci P, Morelli L, Angius A et al: Low-pass DNA sequencing of 1200 Sardinians reconstructs European Y-chromosome phylogeny. Science 2013; 341: 565–569.
Kong A, Frigge ML, Masson G et al: Rate of de novo mutations and the importance of father's age to disease risk. Nature 2012; 488: 471–475.
Kuroki Y, Toyoda A, Noguchi H et al: Comparative analysis of chimpanzee and human Y chromosomes unveils complex evolutionary pathway. Nat Genet 2006; 38: 158–167.
Skaletsky H, Kuroda-Kawaguchi T, Minx PJ et al: The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 2003; 423: 825–837.
Wei W, Ayub Q, Chen Y et al: A calibrated human Y-chromosomal phylogeny based on resequencing. Genome Res 2013; 23: 388–395.
Pink CJ, Swaminathan SK, Dunham I, Rogers J, Ward A, Hurst LD : Evidence that replication-associated mutation alone does not explain between-chromosome differences in substitution rates. Genome Biol Evol 2009; 1: 13–22.
Wilson Sayres MA, Venditti C, Pagel M, Makova KD : Do variations in substitution rates and male mutation bias correlate with life-history traits? A study of 32 mammalian genomes. Evolution 2011; 65: 2800–2815.
Taylor J, Tyekucheva S, Zody M, Chiaromonte F, Makova KD : Strong and weak male mutation bias at different sites in the primate genomes: insights from the human-chimpanzee comparison. Mol Biol Evol 2006; 23: 565–573.
Charlesworth B : The role of background selection in shaping patterns of molecular evolution and variation: evidence from variability on the Drosophila X chromosome. Genetics 2012; 191: 233–246.
Charlesworth B, Charlesworth D : The degeneration of Y chromosomes. Philos Trans R Soc Lond B Biol Sci 2000; 355: 1563–1572.
Wilson MA, Makova KD : Evolution and survival on eutherian sex chromosomes. PLoS Genet 2009; 5: e1000568.
Repping S, Skaletsky H, Brown L et al: Polymorphism for a 1.6-Mb deletion of the human Y chromosome persists through balance between recurrent mutation and haploid selection. Nat Genet 2003; 35: 247–251.
Graves JA : Sex chromosome specialization and degeneration in mammals. Cell 2006; 124: 901–914.
Repping S, van Daalen SK, Korver CM et al: A family of human Y chromosomes has dispersed throughout northern Eurasia despite a 1.8-Mb deletion in the azoospermia factor c region. Genomics 2004; 83: 1046–1052.
Wilson Sayres MA, Lohmueller KE, Nielsen R Natural selection reduced diversity on human Y chromosomes. arXiv preprint:13035012 2013.
King TE, Jobling MA : Founders, drift, and infidelity: the relationship between Y chromosome diversity and patrilineal surnames. Mol Biol Evol 2009; 26: 1093–1102.
Jobling MA : The impact of recent events on human genetic diversity. Philos Trans R Soc Lond B Biol Sci 2012; 367: 793–799.
Chiaroni J, Underhill PA, Cavalli-Sforza LL : Y chromosome diversity, human expansion, drift, and cultural evolution. Proc Natl Acad Sci USA 2009; 106: 20174–20179.
Scally A, Durbin R : Revising the human mutation rate: implications for understanding human evolution. Nat Rev Genet 2012; 13: 745–753.
Miyata T, Hayashida H, Kuma K, Mitsuyasu K, Yasunaga T : Male-driven molecular evolution: a model and nucleotide sequence analysis. Cold Spring Harb Symp Quant Biol 1987; 52: 863–867.
Wyrobek AJ, Eskenazi B, Young S et al: Advancing age has differential effects on DNA damage, chromatin integrity, gene mutations, and aneuploidies in sperm. Proc Natl Acad Sci USA 2006; 103: 9601–9606.
Organisation for Economic Co-operation and Development (OECD). Social Policy Division: SF2.3: Mean age of mothers at first childbirth 2012.
CDC, http://www.cdc.gov/nchs/fastats/births.htm, 2010.
Herlihy D : Medieval Households. Harvard University Press, 1985, pp 104–107.
Parkin TG : Old Age in the Roman World: A Cultural and Social History. JHU Press, 2003, pp 195–196.
Population Division of the Department of Economic and Social Affairs of the United Nations Secretariat: World Population Prospects: The 2012 Revision 2012.
Mendez FL, Watkins JC, Hammer MF : Global genetic variation at OAS1 provides evidence of archaic admixture in Melanesian populations. Mol Biol Evol 2012; 29: 1513–1520.
Mounier Al Marchal Fo, Condemi S : Is Homo heidelbergensis a distinct species? New insight on the Mauer mandible. J Hum Evol 2009; 56: 219–246.
Schwartz JH, Tattersall I : Significance of some previously unrecognized apomorphies in the nasal region of Homo neanderthalensis. Proc Natl Acad Sci USA 1996; 93: 10852–10854.
Bischoff JL, Williams RW, Rosenbauer RJ et al: High-resolution U-series dates from the Sima de los Huesos hominids yields: implications for the evolution of the early Neanderthal lineage. J Archaeol Sci 2007; 34: 763–770.
Finlayson C, Pacheco FG, Rodríguez-Vidal J et al: Late survival of Neanderthals at the southernmost extreme of Europe. Nature 2006; 443: 850–853.
White TD, Asfaw B, DeGusta D et al: Pleistocene Homo sapiens from Middle Awash, Ethiopia. Nature 2003; 423: 742–747.
Clark JD, Beyene Y, WoldeGabriel G et al: Stratigraphic, chronological and behavioural contexts of Pleistocene Homo sapiens from Middle Awash, Ethiopia. Nature 2003; 423: 747–752.
Cann RL, Stoneking M, Wilson AC : Mitochondrial DNA and human evolution. Nature 1987; 325: 31–36.
Dawkins R : The Ancestor's Tale, A Pilgrimage to the Dawn of Evolution. Houghton Mifflin Company: Boston, 2004.
Lachance J, Vernot B, Elbers Clara C et al: Evolutionary history and adaptation from high-coverage whole-genome sequences of diverse African hunter-gatherers. Cell 2012; 150: 457–469.
Yu N, Fu Y-X, Li W-H : DNA polymorphism in a worldwide sample of human X chromosomes. Mol Biol Evol 2002; 19: 2131–2141.
Kaessmann H, Heißig F, Von Haeseler A, Pääbo S : DNA sequence variation in a non-coding region of low recombination on the human X chromosome. Nat Genet 1999; 22: 78–81.
Schuster SC, Miller W, Ratan A et al: Complete Khoisan and Bantu genomes from southern Africa. Nature 2010; 463: 943–947.
Behar DM, van Oven M, Rosset S et al: A ‘Copernican’ reassessment of the human mitochondrial DNA tree from its root. Am J Hum Genet 2012; 90: 675–684.
Acknowledgements
TT was supported by NIH-NICHD: HD070996 and NIH-NIGMS GM068968 grants. We would like to thank Thomas Krahn and Melissa A Wilson Sayres for commenting on this manuscript. We are grateful to two anonymous reviewers who helped us improve the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Elhaik, E., Tatarinova, T., Klyosov, A. et al. The ‘extremely ancient’ chromosome that isn’t: a forensic bioinformatic investigation of Albert Perry’s X-degenerate portion of the Y chromosome. Eur J Hum Genet 22, 1111–1116 (2014). https://doi.org/10.1038/ejhg.2013.303
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.303
Keywords
This article is cited by
-
Between Lake Baikal and the Baltic Sea: genomic history of the gateway to Europe
BMC Genetics (2017)
-
Differential Evolution approach to detect recent admixture
BMC Genomics (2015)
-
Reply to Mendez et al: the ‘extremely ancient’ chromosome that still isn’t
European Journal of Human Genetics (2015)
-
Reply to ‘The ‘extremely ancient’ chromosome that isn’t’ by Elhaik et al
European Journal of Human Genetics (2015)
-
The Y-chromosome point mutation rate in humans
Nature Genetics (2015)
