
Abstract
Recent developments in second-generation sequencing (SGS) technologies provide an avenue for achieving rapid and accurate high-throughput analysis of human and microbial genomic diversity. SGS technologies have the potential to transform existing medical management of complex and life-threatening medical conditions by enabling clinicians to develop disease-targeted clinical care plans for each patient. In this review, we outline how innovative SGS-based approaches can improve the care of recipients of allogeneic hematopoietic cell transplantation (HCT), a life-saving procedure that carries a 1-year mortality risk of over 30%. We specifically evaluate foreseeable applications of SGS-based technology in facilitating rapid, phase-sensitive human leukocyte antigen (HLA) typing, assessment of non-HLA genomic compatibility, identifying patients at high risk for adverse drug reactions, and post-HCT monitoring for engraftment, minimal residual disease and infection. We conclude that innovative SGS approaches have the capacity to revolutionize the HCT recipient risk assessment process, support non-invasive clinical monitoring and improve patient outcomes, thereby setting the stage for a new era of genomically informed patient-centered medicine.
Similar content being viewed by others
Introduction
Approximately 60 000 hematopoietic cell transplantations (HCTs) are performed annually worldwide as the definitive therapy for a wide range of hematological malignancies and non-malignancies.1 More recently, HCT has also been shown to be efficacious in a number of cell-based therapies for non-hematological conditions.2, 3 Once a therapy of last resort for universally lethal diseases, a number of recent scientific and clinical advances, such as the introduction of molecular and sequence-based human leukocyte antigen (HLA) typing, have dramatically improved the long-term success rates of HCT. Despite this progress, the 1-year mortality rate among allogeneic HCT recipients remains a staggering 30–40%, making a potentially life-saving transplant risk-prohibitive for patients with significant comorbidities, at advanced ages, or for whom no appropriate HLA-matched donors can be identified.4
The degree of HLA matching between the donor and the recipient strongly determines the recipient’s risk for graft-versus-host disease (GVHD), which alongside infection and disease relapse are the three major causes of transplant-related mortality (TRM).5, 6 Unlike HLA matching, which is largely standardized across HLA laboratories at major transplant centers, other clinical aspects of HCT management, including pre-conditioning chemotherapy, prophylactic or therapeutic antibiotic use and disease-relapse monitoring, vary significantly from center to center and by clinician experiences.
Integrating histocompatibility (including HLA) matching and other genomic markers for GVHD into cumulative risk models specific for a given patient’s diagnosis, disease severity, and demographics offers a method for testing and establishing quantitative, evidence-based risk prediction algorithms. In the last 5 years, a number of prospective and retrospective analyses have reported the evaluation of combinations of genetic and clinical data (aside from HLA matching), in TRM/GVHD risk assessment. However, a clinically validated risk assessment algorithm that fully utilizes the available genomic data has not yet been developed or tested.7, 8 While it may not always be possible to improve donor–recipient matching, as available donors are limited, quantitative, patient-specific risk scores can be useful in the clinical care setting. Accurate assessment and quantification of the underlying risk in relation to the frequency of monitoring of GVHD, infection risk and relapse can have major impact on HCT outcome and overall survival.
In the last decade, developments in the field of genomics, particularly the recent breakthroughs in second-generation sequencing (SGS)-based technologies, have stimulated the development of individual patient-based therapeutic approaches and methods for rapid and precise genetic testing. However, the full potential of these advances in directing patient care are far from realized in the field of HCT. We present, in this review, a vision for how some of the forefront SGS technologies and approaches can improve the assessment of pre-transplant risk, individualize pharmacotherapy and inform long-term disease relapse monitoring (Figure 1). Although our review focuses on SGS applications to HCT, this is only one of many evolving models of how emerging genomics tools are empowered to deliver individualized patient therapy. As the technical aspects of most of the SGS technologies discussed here are expansive, we provide a brief summary and references for a wide range of SGS-based technologies that have been well-described elsewhere (see the technologies detailed in Table 1 and a brief overview of them in Box 1).

Clinical applications of second-generation sequencing (SGS) technologies to HCT. (a) Diagram of typical workflow from clinical sample to sequencing. SGS of genomic DNA derived from recipient somatic or tumor-cell DNA can be used to profile genomic risk factors of underlying disease or identify causative driver mutations in hematological malignancies. Although the exact methods are platform specific, typical SGS-based techniques follow a standard workflow, requiring the isolation of genomic DNA, DNA fragmentation and the ligation of paired-end adaptors. Depending on whether the whole genome, all coding or specific coding regions are interrogated, additional sequence-specific adaptors for exonic or user-specified regions are used to capture the prespecified target DNA fragments. Adapter-ligated fragments are next captured either in an oil-emulsion bead (454 pyrosequencing) or on a solid platform (bridge amplification) as shown here. All SGS techniques culminate with massively parallel sequencing technology utilizing an array-like platform. As nucleotide bases are added cyclically, light is produced and sequences are read out based on detected fluorescence intensities and wavelengths. (b) Existing and potential SGS applications to HCT patient care using available SGS technology: As discussed in the main text, SGS-based improvements to existing clinical care of HCT recipients can be broadly classified into three major domains: (1) accurate and rapid HLA typing, (2) pre-transplant assessment of adverse drug response and non-HLA genomic risk factors and (3) post-HCT early and routine monitoring for minimal residual disease, engraftment of donor cells, and life-threatening systemic infections.
SGS applications to HLA matching
The HLA class I (A, B and C) and class II (DP, DQ and DR) molecules, encoded within the MHC (Major Histocompatibility Complex) on chr6p21, are codominantly expressed, cell-surface receptors that serve dual functions. The HLAs, encoding the variable chains of the MHC receptors, present self and foreign peptide antigens to helper and cytotoxic T cells, and as a combination, make up a set of ‘self-identifying’ markers that are unique to a given individual. The clinical gold standard for identifying two individuals with matching HLAs at the amino-acid level, also known as high-resolution HLA matching, relies on either Sanger sequencing or other molecular tests to compare the genotypes at each of the eight (HLA-A, B, C, and DRB1) or ten (HLA-A, B, C, DQB1 and DRB1) highly polymorphic loci.9 An 8/8 HLA-matched HCT refers to a donor–recipient pair matched for HLA-A, B, C, and DRB1, whereas a 10/10 match also includes the HLA-DQB1 locus.
While Class I HLAs are highly expressed across most somatic cells, Class II HLAs are predominantly expressed on the surface of professional antigen-presenting immune cells. Furthermore, while Class I antigens are composed of a polymorphic alpha chain and an invariable beta chain (encoded by a non-MHC gene beta-2-microglobulin), Class II antigens are heterodimers composed of MHC-encoded alpha and beta chains, though the beta chains are typically more variable in sequence and consequently genotyped during clinical HLA matching. While HLA matching is critical for the success of allogeneic HCT, the degree to which a given mismatch will negatively predict outcome may vary depending on the antigen type, the relative antigenicity and expression level of a given HLA allele, as well as the underlying disease process and pre-conditioning regimen. Definitive and quantitative studies that assess these risk prediction models are still needed.
The ‘antigenic’ or hypervariable domains of the polymorphic chain of each HLA are accessible and recognized by corresponding T cells; the extreme diversity of the HLA repertoire in humans is, in part, attributable to the recent positive selection of this region in the human genome. Over the course of normal development, sequential processes of positive and negative selection enable T cells to accommodate the unique HLA repertoire of each individual, thereby facilitating self-tolerance. However, in the context of HCT, when donor T cells are introduced to the tissues of a ‘foreign’ recipient, any mismatching within these polymorphic regions will likely be strongly immunogenic, often irrespective of the specifically bound antigen. This consequently may elicit cross-reactive donor T cells to mount an immune response to host tissues that clinically manifests as GVHD.
Acute GVHD (aGVHD), occurring within 100 days after transplant, can cause local inflammation to the skin, liver and gastrointestinal tract,10 but can also result in systemic tissue damage that is life threatening. The strongest predictor of aGVHD risk is the degree of HLA mismatching between the donor and the recipient,5 making the identification of an HLA-matched donor critical for the control and prevention of aGVHD. Compared with 8/8 HLA-matched HCT recipients, the risk of developing aGVHD is higher for one or two HLA-mismatched recipients.5, 9 Since sequence-based HLA typing for donor selection became the standard of care at most transplant centers, short-term TRM has dramatically declined.6 Recent reports from the National Marrow Donor Program show that outcomes for HLA-matched, unrelated donor HCTs (MUD-HCTs) are now comparable to that of HLA-matched related donor HCTs (MRD-HCT), underscoring the critical importance of HLA matching in HCT outcomes.6
However, GVHD remains a major cause of morbidity and mortality, even after 8/8 and 10/10 MRD and MUD-HCTs, suggesting that non-HLA-A, B, C, DQB1 and DRB1 genetic differences may be important in allorecognition. For example, HLA-DP, which is poorly expressed on the surface of lymphocytes, was not historically considered in matching.11 However, in a 2012 retrospective analysis of nearly 12 000 HCTs using high-resolution HLA typing, Petersdorf et al showed that HCT transplants with HLA-DPB1 non-permissive mismatching, as defined by T-cell epitope cross-reactivity groups, are significantly associated with poorer TRM in 10/10 and 9/10 MUD-HCT.12 Similar studies on other classically ‘low-expression’ HLAs (including HLA-C, DQ and DR51/52/53) have also recently been shown to affect transplant outcome.13, 14 These findings underscore the value of large-scale longitudinal analysis, the use of molecular and sequence-based HLA typing, and the increasing importance of non-classical HLA genes in HCT outcome as most allogeneic HCTs are now fully or near-fully HLA matched.
Under existing methods, typing additional HLA loci requires significant increases in time, effort and costs. However, a recent multi-center study demonstrated the efficiency and feasibility of highly accurate SGS-based HLA typing,15 showing improved accuracy, coverage and turn-around times of SGS at a comparable cost to lower resolution probe-based tests.16 Another drawback of existing methods is that most analyze only the highly variable exons of the typed gene to minimize cost. Consequently, non-synonymous single-nucleotide variants (nsSNVs) outside these targeted regions are missed. SGS makes targeted sequencing of entire HLA genes feasible with minimal increase in cost.17 In light of the recent findings that HLA-DP and other non-classical HLA mismatches also correlate with increased risk of TRM and GVHD, additional genetic information captured with SGS-based HLA typing may have clinical utility, particularly where multiple equivocally matched donors are identified based on classical HLA typing.
Finally, reflex testing is often required to resolve typing ambiguities that arise from Sanger sequencing, which does not yield phase information for heterozygous sites. In contrast, SGS-based HLA typing using tiled, paired-end primer designs accurately assess MHC haplotype phasing, thereby reducing the frequency of typing ambiguities and the need to perform reflex testing. As the throughput and read length of SGS continue to improve, soon it will be feasible to sequence the entire MHC in a few reads in just a few hours, eliminating time delay as a barrier to the use of HLA matching data to inform the treatment of solid organ transplant (SOT) recipients (eg, in pre-transplant immunosuppressive dosing).18
Application of SGS technology to GVHD risk assessment beyond the HLA
As the relative number of 9/10 and 10/10 MUD-HCTs performed have increased compared with less well-matched HCTs, it is becoming more clear that non-HLA genetic mismatching also underpin GVHD and overall TRM. For example, pediatric acute lymphoblastic leukemia (ALL) patients receiving MUD-HCT have a 2.42- and 5.12-fold increased risk of acute and chronic GVHD, respectively, compared with recipients of HLA-identical sibling donor transplants.9 Since donors and recipients from a MUD-HCT are HLA-matched by state, rather than by descent, the higher observed risk of GVHD among MUD-HCT versus MRD-HCT recipients could be attributable to non-HLA-A, B, C, and DRB1 genetic disparities within and beyond the MHC.
In addition, Terasaki19 reported in a large retrospective analysis of rejection data from SOT that HLA mismatch accounted for just 18% of rejection risk, while 38% is attributable to non-HLA differences in genetic compatibility. In fact, sources of human genomic variations aside from the classical HLAs including non-synonymous SNPs (nsSNPs), copy number variations (CNVs) and nucleotide insertion/deletions (indels) in genes encoding cell-surface proteins are known to contribute to the ‘non-HLA’ histocompatibility repertoire.20 These include genes that encode the Killer-cell Immunoglobulin-like Receptor family members (KIRs), MIC-A and MIC-B antigens,21 and minor histocompatibility antigens (mHAs).
The mHAs, a group of polymorphic peptides that can be presented by specific MHC molecules to mediate T-cell allorecognition in the context of HLA-matched transplants, are encoded as nsSNPs or other genetic variations across ∼50 (as of 2012) autosomal mHA genes.1 In addition, there are Y-chromosomally encoded mHAs (H-Y antigens), for which mismatching has been shown to increase the GVHD and TRM risk in recipients of female-to-male MRD-HCTs (F-to-M), as compared with non-F-to-M recipients.22, 23
That only a few dozen mHAs are known is surprising given the sequence diversity of the human genome. Depending on ancestry and relatedness, two humans have ∼3–10 million divergent variants, or one base-pair difference per 300–1000 bases. As 3–5% of these polymorphisms are in coding regions of the genome, many more mHAs may exist but have remained unrecognized.24 As mHAs were historically identified using biochemical means, state of the art genomic or proteomic screens may reveal the existence of many more mHAs.25
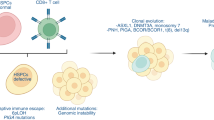
Recent efforts using high-throughput genomics, in-silico prediction and candidate gene resequencing have already yielded some successes,26 for example, in the recent discovery of histocompatibility genes that occur as a result of functional gene expression knockout by McCarroll et al.27 Such genes encode gene-expressed mismatch antigens (GEMAs), which are not expressed in a significant percentage of the human population due to the presence of two non-complementary null mutations at a single locus (Figure 2). In the context of transplantation, when donors and recipients are mismatched in the expression of a GEMA, GEMA-derived peptides are antigenic in the GEMA(−) individual. Analogous to the role of H-Y antigens in F-to-M HCT transplants, GEMA mismatch can thus cause GVHD in HCT recipients if GEMA(−) donor lymphocytes are transplanted to a recipient whose somatic cells are GEMA(+).

The role of gene-expressed mismatch antigens in GVHD or rejection following transplantation. (a) Donor-recipient GEMA mismatch in myeloablative HCT: GVHD can occur in an HCT recipient with normal GEMA expression when he or she is transplanted with cells derived from a donor without normal GEMA protein expression. Since GEMAs are ‘self-expressed’ antigens, GEMA-derived peptides can be presented in the context of both class I and class II MHC receptors found on recipient cells following HCT. Allorecognition of host MHC molecules presenting GEMA-derived peptides subsequently triggers donor-derived T cells that are not ‘tolerized’ to GEMA-derived peptides to attack host cells, resulting in clinical manifestations of GVHD. In addition, because GEMAs such as UGT2B17 can be cell-surface antigens, their antigenicity is not MHC-restricted like most autosomally encoded minor histocompatibility antigens. Like H-Y antigens, a B-cell mediated response to the ‘foreign’ GEMA expression can result in the production of autoantibodies by donor-derived B cells that target the host-cell expressed GEMA proteins on the cell surface. McCarroll et al. first demonstrated the clinical significance of GEMA mismatch in HCT by following up on the results from a large genome-wide association study (GWAS)-based screen for homozygous deletion copy number variations (hdCNVs) that appear to be tolerated and are found frequently in an appreciable percentage of the human population. Based on the hypothesis that hdCNVs overlapping a gene-encoding region would cause gene expression mismatch between HCT donors and recipients, McCarroll screened ∼400 HCT recipients and their HLA-matched siblings for six candidate GEMAs. They identified that D−/R+ status for one particular GEMA UGT2B17 increased the risk of Grade 2+ GVHD, a finding that was later replicated in two independent cohorts, together totaling over 1300 patients (combined OR of 2.5).18 In addition, McCarroll et al showed that UGT2B17+ HCT recipients, who received hematopoeitic cells from UGT2B17−/− donors were seropositive for anti-UGT2B17 antibodies. Moreover, they were able to isolate MHC-bound UGT2B17-derived peptides from the sera of these UGT2B17 mismatched. Given that the average human genome has at least several thousand CNVs greater than 1 kb (which is likely to be an underestimate due to current limitations in CNV detection platforms19) and contains ∼100 loss of function (LoF) mutations (with ∼20 in both copies) 20, other GEMAs are likely to exist. By coupling large-scale RNA-seq and SGS data, a genome-wide search for other GEMAs may reveal hundreds of other such loci across the genome. (b) Donor-recipient GEMA mismatch in the context of reduced-intensity conditioning/non-myeloablative HCT or solid organ transplantation: rejection of donor cells or tissue occurs due to the absence of GEMA protein expression (eg, UGT2B17) in the recipient. Complementary to the scenario illustrated in (a), recipient tissues do not express GEMA proteins. Thus, potentially GEMA-reactive T- and B-cell clones are not eliminated from the host immune repertoire. Following transplantation, immune competent recipient T cells can attack donor stem cells expressing GEMA and inhibit engraftment or cause acute graft rejection.
Identifying GEMAs and other genomic loci that contribute to allogenicity may also identify donor–recipient genetic disparities that are beneficial, such as the KIRs, which are encoded by a cluster of highly polymorphic and copy number variable genes at chr19q13.4.21, 28 KIRs negatively regulate the activity of NK cells when they are stimulated by their cognate ligands, the NK-inhibitory ligands encoded within the MHC. KIR mismatching in the context of MRD-HCTs or MUD-HCTs results in the disinhibition of KIRs, since the donor NK cells are not suppressed by the ligands expressed in a KIR-mismatched recipient. The disinhibition of NK cell activity appears to improve survival among patients receiving KIR-mismatched, HLA-matched HCTs for lympho- and myeloproliferative diseases by boosting the graft-versus-leukemia (GVL) effect.29, 30 As the genetic inheritance of KIRs and the MHC loci are independent meiotic events, on average 75% of MRD-HCT and nearly all MUD-HCTs are KIR mismatched. Other GVHD-protective genomic markers may exist with similar or disparate mechanisms, and identifying them may benefit from creative applications of unbiased genomic studies among large MRD/MUD-HCT cohorts. However, such non-HLA effects, albeit clinically significant, may be minor relative to the effect of HLA, or could be HLA-dependent.
Although the use of non-HLA factors in donor selection may not be practical given the limited donor availability, identification of polymorphisms in mHAs, GEMAs or immunoregulatory genes that are important for HCT outcome can be used to develop a multi-factorial GVHD and TRM risk assessment model.7, 26, 31 Such a model will need to differentially weigh HLA and non-HLA factors based on their ability to predict the likelihood and severity of TRM and GVHD. As allele-level HLA typing and non-HLA genetic data are now cataloged for patients across large allogeneic HCT recipient databases, multi-variate risk prediction algorithms developed using complex and large-scale genomic data may enable the development of risk-prediction algorithms. Definitive evaluation of clinical implications of these findings will require careful analysis of results from large randomized, clinical trials that test the utility of prospective risk-stratification efforts.
Application of pharmacogenomics (PGx) to HCT
Dozens of GWAS and RNA expression studies published in the last decade have identified dozens of significant associations between the presence of genetic polymorphisms and changes in gene expression to the risk of adverse drug response (ADR), respectively. For example, ∼1 in 300 individuals have complete LoFs (null mutations on two copies) in the gene encoding thiopurine methyltransferase (TPMT), the hepatic enzyme required for 6-Mercaptopurine (6-MP) degradation. Administering standard 6-MP doses to ALL patients with underlying TPMT deficiency causes life-threatening ADR.32 Currently, TPMT genotyping can be routinely ordered before chemotherapy to identify those at risk for ADR, but also to identify heterozygous TPMT mutation carriers, who show improved response rates to 6-MP treatment compared with patients with two normal copies of TPMT.33
In addition to TPMT and other-characterized examples of ADR, studies applying large-scale SGS and GWAS screening methods have identified a number of rarer mutations in proteins either directly or indirectly involved in drug metabolism or enzyme synthesis that increase the risk of ADR or significantly affect therapeutic outcomes. Toward this end, GWAS has been particularly successful in identifying pharmacogenetic (PGx) loci by SNP genotyping and expression quantitative trait loci (eQTL) analysis. Notable examples include mutations in Interleukin-15 (IL15) associated with altered disposition of antileukemic chemotherapy (pediatric ALL), as well as those in CYP2C9 associated with bisphosphonate-induced jaw osteonecrosis (multiple myeloma), among others34, 35, 36 (see Table 2 for a detailed list of known PGx loci). In part, the success of GWAS is related to the fact that many ADR-associated mutations are relatively common in the population, since these ‘evoked’ phenotypes are otherwise not deleterious, such mutations are typically not under negative selection pressure.
By enabling clinicians to make informed, patient-specific therapeutic choices that favor therapeutic response while minimizing ADRs, PGx knowledge may lead to a significant life and cost-savings. Approximately 20% of all injuries and deaths among hospitalized patients in the United States has been attributed to ADRs,37 incurring an annual event-related health-care cost of over 136 Billion USD.38 Since the rate of ADRs rises exponentially among patients receiving four or more medications,39 evaluating and minimizing ADR risk is particularly important for HCT recipients, who are exposed to high doses of multi-drug combinations over the course of therapy.
An excellent implementation of PGx in direct patient care is exemplified by the electronic Medical Records and Genomics (eMERGE) Network pilot program, which integrates the results of capture amplicon sequencing (CAS), a form of targeted SGS, of 84 key PGx genes into patient electronic medical records (EMRs) that are directly accessible to treating physicians. This initiative is part of an evolving effort to improve the portability and accessibility of genomic information to facilitate individualized patient care, in the hope of improving long-term and peri-operative ADR risk management.40 By limiting the total number of loci interrogated, CAS allows panel-based multiplex genetic testing of selective loci at high-throughput with improved cost efficiency. CAS is particularly suited as a clinical tool for the assessment of a set of informative genes, such as enzymes that act in drug metabolism. In the context of HCT, a CAS panel developed for pre-transplant risk assessment that includes PGx and non-MHC histocompatibility loci may be a viable option until WGS and ES become available economically on a large scale.
SGS applications in post-transplant monitoring
Sensitive detection of donor–recipient cellular chimerism, minimal residual disease (MRD), and early stages of microbial infections are critical to post-HCT clinical care, particularly in patients with underlying malignancy. Existing surveillance methods aimed at early detection of cancer cells in post-HCT chimeric hematopoietic systems rely on STR typing with an ∼2–3% detection limit. Several groups have reported the successful application of SGS technology to achieve improved detection sensitivity for low copies of genomic DNA that are the minor component in mixed cellular or acellular samples, including sera.
Compared with molecular and Sanger sequencing-based methods, SGS is sensitive enough to detect mutations in single tumor cells,41, 42 which is the ultimate goal of MRD detection. For example, CAS-based profiling of total genomic DNA isolated from the peripheral blood of breast and ovarian cancer patients has been used for routine monitoring of patients who achieved disease remission. This non-invasive method has been shown to detect TP53 (P53) and EGFR mutations during early stages of cancer relapse by detecting rare cancer-specific polymorphisms (MAF∼2%) at sensitivities and specificities >97%.43 SGS has also been used clinically to diagnose hematopoietic malignancies and inform cancer immunotherapy. Another study has shown that exonic mutations in a select set of cancer genes, including TP53 and RUNX1, improved prognostic significance in 439 patients with myelodysplastic syndrome.44 Coupling single-cell isolation methods such as flow cytometry with CAS or WGS could enable MRD monitoring at sensitivities and specificities beyond that achievable by existing methods.42 For instance, Wu et al45 have shown, using SGS of variable regions of T-cell Receptor Beta and Gamma (TCRB and TCRG) genes of patients with T-lineage ALL, superior detection of early disease relapse compared with established flow-based methods.
Furthermore, SGS methods have been applied to the management of post-transplant recipients, including the non-invasive detection of acute cellular rejection following heart transplantation. By monitoring the levels of donor genomic DNA levels in the sera of recipients, Synder et al46 accurately predicted if the recipient is experiencing early stages of acute graft rejection. Others have shown that combined PCR and SGS quantification of T-cell repertoire recovery can successfully predict which patients are at high risk of infection or disease relapse following HCT.47
Finally, early detection and therapy of microbial infections are crucial determinants of HCT outcomes as >30% of HCT recipients develop an episode of bacteremia in the first weeks following transplant and 15% of all mortalities following unrelated allogeneic HCT are infection related.5 Rapid and accurate detection, identification and drug-susceptibility phenotyping methods for isolated clinical pathogen(s) using WGS and CAS of non-human DNA or RNA48, 49 using genomic or ribosomal multi-locus sequence typing (MLST/rMLST) already exist. In one approach, for example, a CAS panel of 53 bacterial genes that encode the bacterial ribosome protein subunits uniquely catalogs >1900 bacterial genomes from 452 bacterial genera. Sequences derived from clinical isolates can then be queried against this database for rapid strain-level identification.50
Unlike RT-PCR or array platforms, SGS detects a wide range of pathogens in a single test, eliminating single pathogen tests. This method is particularly helpful for the early detection and typing of fungal infections, which remain a major cause of morbidity and mortality in immunocompromised HCT recipients. With few exceptions, fungal typing requires the culture of slow-growing organisms, making rapid diagnosis within an already narrow therapeutic time-window challenging (eg, Mucor).51 Although SGS of clinical microbial samples currently requires an intermediate culture and isolation step, metagenomic approaches using third-generation sequencing (TGS) are currently being developed. These TGS platforms do not require amplification or culturing, and affords single DNA molecule sequencing of samples directly acquired from body fluids or sample sites.49, 52
In addition, SGS can be coupled with PGx to improve antimicrobial selection and dosing by detecting high-resistance strains that require strain-specific therapy and reducing the frequency of severe ADRs to anti-microbial agents. SGS has already transformed clinical practices in the care of highly virulent infections such as HIV. Over 300 000 HIV-1 strains have been sequenced worldwide and matched with drug-susceptibility data, providing a large electronic database that matches HIV DNA sequences of a patient-specific infection to drug susceptibility patterns. This resource enables physicians to prescribe individualized HAART (highly active anti-retroviral therapy) regimens for patients based on the drug resistance pattern predicted for their virus.53 A similar strategy has also been efficacious in using SGS to identify MRSA, influenza, or specific pathogenic E. coli strains from related, but benign strains.54, 55
From research findings to clinical deliverables
Since the first draft human genome was completed, requiring $3 billion USD over 13 years, the technological and scientific advances that have been made in human genetics have exponentially accelerated. Indeed, SGS can now accomplish the equivalent task in <30 h for the cost of a magnetic resonance imaging scan (∼2–3000 USD).18, 56 Although the clinical implementation of SGS technology to routine patient care will require significant upfront costs in equipment acquisition and personnel training, these investments will be worthwhile. In fact, three companies have released bench-top SGS instruments intended to reduce the cost of SGS technology and to increase their accessibility at smaller institutions.18 These portable technologies can sequence an entire human genome (100 × the size of that of the average E. coli) in just 15 min to 3 h.57 Such a timeframe makes the use of SGS for infection or MRD monitoring feasible over the course of a clinic visit or on the hospital floor.
Taken together, SGS can be used to provide pre-HCT histocompatibility, ADR and GVHD risk assessment. As illustrated in Figure 3, the use of CAS or WGS as a part of post-HCT care also enables high-sensitivity engraftment assessment, infection detection and disease relapse monitoring. Although the expense of SGS methods is not trivial, a single allogeneic HCT and peri-procedural care cost on average over 200 000 USD; in comparison, the cost of WGS is fast approaching 1% of this sum.58 The real challenge is thus not necessarily cost, but rather in determining how to streamline SGS-based methods for clinical applications and concretely defining how genomic information should be appropriately used in the patient-care setting.

The application of genomics approaches to improve HCT outcomes. (Left) panels show existing clinical practice, (Center) panel describes recent scientific/research contributions through genomic technology and (Right) panel proposes clinically feasible applications of genomics technology in the coming years. The four categories correspond to the four areas in which genomics technology will likely make the most direct impact on the clinical management of HCT recipients.
In the coming years, physicians can use SGS to prospectively acquire genomics information about a recipient and potential donor(s), and thereby, refine donor choice, assess GVHD risk and facilitate individualized chemotherapy. Post-HCT applications of SGS can expedite MRD monitoring and infection diagnosis. Together, this suite of advances in HCT recipient management exemplifies the versatility of SGS-based genomics applications in delivering personalized, evidence-based patient care.
References
BMDW: Bone Marrow Donors Worldwide Annual Report 2011. Leiden: The Netherlands Europdonor Foundation, 2012.
Kiem H-P, Jerome KR, Deeks SG, McCune JM : Hematopoietic-stem-cell-based gene therapy for HIV disease. Cell Stem Cell 2012; 10: 137–147.
Daley GQ, Scadden DT : Prospects for stem cell-based therapy. Cell 2008; 132: 544–548.
Zeiser R, Marks R, Bertz H, Finke J : Immunopathogenesis of acute graft-versus-host disease: implications for novel preventive and therapeutic strategies. Ann Hematol 2004; 83: 551–565.
Pasquini M, Wang Z : Current use and outcome of hematopoietic stem cell transplantation. CIBMTR Summary Slides 2011.
Spellman SR, Eapen M, Logan BR et al: A perspective on the selection of unrelated donors and cord blood units for transplantation. Blood 2012; 120: 259–265.
Chien JW, Zhang XC, Fan W et al: Evaluation of published single nucleotide polymorphisms associated with acute GVHD. Blood 2012; 119: 5311–5319.
Hansen JA, Chien JW, Warren EH, Zhao LP, Martin PJ : Defining genetic risk for graft-versus-host disease and mortality following allogeneic hematopoietic stem cell transplantation. Curr Opin Hematol 2010; 17: 483–492.
Zhang M-J, Davies SM, Camitta BM, Logan B, Tiedemann K, Eapen M : Comparison of outcomes after HLA-matched sibling and unrelated donor transplantation for children with high-risk acute lymphoblastic leukemia. Biol Blood Marrow Transplant 2012; 18: 1204–1210.
Benesch M, Deeg HJ in Soiffer RJ (ed.):: Acute Graft-versus-Host Disease: Hematopoietic Stem Cell Transplantation. Totowa, NJ: Humana Press, 2008, pp 589–620.
Lee SJ, Klein J, Haagenson M et al: High-resolution donor-recipient HLA matching contributes to the success of unrelated donor marrow transplantation. Blood 2007; 110: 4576–4583.
Fleischhauer K, Shaw BE, Gooley T et al: Effect of T-cell-epitope matching at HLA-DPB1 in recipients of unrelated-donor haemopoietic-cell transplantation: a retrospective study. Lancet Oncol 2012; 13: 366–374.
Woolfrey A, Klein JP, Haagenson M et al: HLA-C antigen mismatch is associated with worse outcome in unrelated donor peripheral blood stem cell transplantation. Biol Blood Marrow Transplant 2011; 17: 885–892.
Fernández MA, Klein JP, Haagenson M et al: Multiple mismatches at the low expression HLA loci DP, DQ, and DRB3/4/5 associate with adverse outcomes in hematopoietic stem cell transplantation. Blood 2013; 121: 4603–4610.
Holcomb CL, Höglund B, Anderson MW et al: A multi-site study using high-resolution HLA genotyping by next generation sequencing. Tissue Antigens 2011; 77: 206–217.
Wang C, Krishnakumar S, Wilhelmy J et al: High-throughput, high-fidelity HLA genotyping with deep sequencing. Proc Natl Acad Sci 2012; 109: 8676–8681.
Lind C, Ferriola D, Mackiewicz K et al: Next-generation sequencing: the solution for high-resolution, unambiguous human leukocyte antigen typing. Hum Immunol 2010; 71: 1033–1042.
Loman NJ, Misra RV, Dallman TJ et al: Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotech 2012; 30: 434–439.
Terasaki P : Deduction of the fraction of immunologic and non-immunologic failure in cadaver donor transplants. Clin Transpl 2003; 449–452.
Armour JAL : Copy number variation and antigenic repertoire. Nat Genet 2009; 41: 1263–1264.
Stephens HAF : MICA and MICB genes: can the enigma of their polymorphism be resolved? Trends Immunol 2001; 22: 378–385.
Kim Y-H, Faaij CMJM, van Halteren AGS et al: In situ detection of HY-specific T cells in acute graft-versus-host disease-affected male skin after sex-mismatched stem cell transplantation. Biol Blood Marrow Transplant 2012; 18: 381–387.
Nannya Y, Kataoka K, Hangaishi A, Imai Y, Takahashi T, Kurokawa M : The negative impact of female donor/male recipient combination in allogeneic hematopoietic stem cell transplantation depends on disease risk. Transplant Int 2011; 24: 469–476.
Jorde LB, Wooding SP : Genetic variation, classification and 'race'. Nat Genet 2004; 36 (11 Suppl): S28–S33.
DeLuca DS, Eiz-Vesper B, Ladas N, Khattab BA-M, Blasczyk R : High-throughput minor histocompatibility antigen prediction. Bioinformatics 2009; 25: 2411–2417.
Warren EH, Zhang XC, Li S et al: Effect of MHC and non-MHC donor/recipient genetic disparity on the outcome of allogeneic HCT. Blood 2012; 120 (14): 2796–2806.
McCarroll SA, Bradner JE, Turpeinen H et al: Donor-recipient mismatch for common gene deletion polymorphisms in graft-versus-host disease. Nat Genet 2009; 41: 1341–1344.
Ruggeri L, Mancusi A, Burchielli E et al: Natural killer cell recognition of missing self and haploidentical hematopoietic transplantation. Semin Cancer Biol 2006; 16: 404–411.
Miller JS, Blazar BR : Control of acute myeloid leukemia relapse — dance between KIRs and HLA. N Engl J Med 2012; 367: 866–868.
Ruggeri L, Capanni M, Urbani E et al: Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002; 295: 2097–2100.
Spierings E, Kim Y-H, Hendriks M et al: Multicenter analyses demonstrate significant clinical effects of minor histocompatibility antigens on GvHD and GvL after HLA-matched related and unrelated hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013; 19 (8): 1244–1253.
Carroll WL, Bhojwani D, Min D-J et al: Pediatric acute lymphoblastic leukemia. ASH Education Program Book 2003; 2003: 102–131.
Relling MV, Hancock ML, Boyett JM, Pui C-H, Evans WE : Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 1999; 93: 2817–2823.
Cheok MH, Pottier N, Kager L, Evans WE : Pharmacogenetics in acute lymphoblastic leukemia. Semin Hematol 2009; 46: 39–51.
Yang JJ, Cheng C, Yang W et al: Genome-wide interrogation of germline genetic variation associated with treatment response in childhood acute lymphoblastic leukemia. JAMA 2009; 301: 393–403.
Paugh SW, Stocco G, McCorkle JR, Diouf B, Crews KR, Evans WE : Cancer pharmacogenomics. Clin Pharmacol Ther 2011; 90: 461–466.
Leape LL, Brennan TA, Laird N et al: The nature of adverse events in hospitalized patients. N Engl J Med 1991; 324: 377–384.
Johnson Ja, Bootman JL : Drug-related morbidity and mortality: a cost-of-illness model. Arch Intern Med 1995; 155: 1949–1956.
Jacubeit T, Drisch D., Weber E. : Risk factors as reflected by an intensive drug monitoring system. Agents Actions 1990; 29: 117–125.
Gottesman O, Kuivaniemi H, Tromp G et al: The Electronic Medical Records and Genomics (eMERGE) Network: past, present, and future. Genet Med 2013; 15: 761–771.
Zong C, Lu S, Chapman AR, Xie XS : Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 2012; 338: 1622–1626.
Navin N, Hicks J : Future medical applications of single-cell sequencing in cancer. Genome Med 2011; 3: 31.
Forshew T, Murtaza M, Parkinson C et al: Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med 2012; 4: 136ra168.
Bejar R, Stevenson K, Abdel-Wahab O et al: Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 2011; 364: 2496–2506.
Wu D, Sherwood A, Fromm JR et al: High-throughput sequencing detects minimal residual disease in acute T lymphoblastic leukemia. Sci Transl Med 2012; 4: 134ra163.
Snyder TM, Khush KK, Valantine HA, Quake SR : Universal noninvasive detection of solid organ transplant rejection. Proc Natl Acad Sci 2011; 108: 6229–6234.
van Heijst JWJ, Ceberio I, Lipuma LB et al: Quantitative assessment of T cell repertoire recovery after hematopoietic stem cell transplantation. Nat Med 2013; 19: 372–377.
Bille E, Zahar J-R, Perrin A et al: A chromosomally integrated bacteriophage in invasive meningococci. J Exp Med 2005; 201: 1905–1913.
Didelot X, Bowden R, Wilson DJ, Peto TEA, Crook DW : Transforming clinical microbiology with bacterial genome sequencing. Nat Rev Genet 2012; 13: 601–612.
Jolley KA, Bliss CM, Bennett JS et al: Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology 2012; 158: 1005–1015.
Cumbo TA, Segal BH : Prevention, diagnosis, and treatment of invasive fungal infections in patients with cancer and neutropenia. J Natl Compr Cancer Netw 2004; 2: 455–469.
Schadt EE, Turner S, Kasarskis A : A window into third-generation sequencing. Human Mol Genet 2010; 19: R227–R240.
Relman DA : Microbial genomics and infectious diseases. N Engl J Med 2011; 365: 347–357.
Köser CU, Holden MTG, Ellington MJ et al: Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N Engl J Med 2012; 366: 2267–2275.
Rasko DA, Rosovitz MJ, Myers GSA et al: The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J Bacteriol 2008; 190: 6881–6893.
Jiang Q, Turner T, Sosa MX, Rakha A, Arnold S, Chakravarti A : Rapid and efficient human mutation detection using a bench-top next-generation DNA sequencer. Hum Mutat 2012; 33: 281–289.
Check Hayden E : Nanopore genome sequencer makes its debut. Nature (2012), doi:10.1038/nature.2012.10051.
Blommestein HM, Verelst SGR, Huijgens PC, Blijlevens NMA, Cornelissen JJ, Uyl-de Groot CA : Real-world costs of autologous and allogeneic stem cell transplantations for haematological diseases: a multicentre study. Ann Hematol 2012; 91: 1945–1952.
Acknowledgements
YRL is supported by the NIH Medical Scientist Training Program and the Paul and Daisy Soros Fellowship for New Americans. This work was supported in part by the NHGRI (eMERGE) grants U01HG006830 and 3U01HG006830-S1.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Li, Y., Levine, J., Hakonarson, H. et al. Making the genomic leap in HCT: application of second-generation sequencing to clinical advances in hematopoietic cell transplantation. Eur J Hum Genet 22, 715–723 (2014). https://doi.org/10.1038/ejhg.2013.247
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.247
