
Abstract
The three-base-pair deletion c.1521_1523delCTT (p.Phe508del, F508del) in the cystic fibrosis transmembrane conductance regulator (CFTR) is the most frequent disease-causing lesion in cystic fibrosis (CF). The CFTR gene encodes a chloride and bicarbonate channel at the apical membrane of epithelial cells. Altered ion transport of CFTR-expressing epithelia can be used to differentiate manifestations of the so-called CF basic defect. Recently, an 11p13 region has been described as a CF modifier by the North American CF Genetic Modifier Study Consortium. Selecting the epithelial-specific transcription factor EHF (ets homologous factor) as the likely candidate gene on 11p13, we have genotyped two intragenic microsatellites in EHF to replicate the 11p13 finding in the patient cohort of the European CF Twin and Sibling Study. We could observe an association of rare EHF haplotypes among homozygotes for c.1521_1523delCTT in CFTR, which exhibit a CF-untypical manifestation of the CF basic defect such as CFTR-mediated residual chloride secretion and low response to amiloride. We have reviewed transcriptome data obtained from intestinal epithelial samples of homozygotes for c.1521_1523delCTT in CFTR, which were stratified for their EHF genetic background. Transcripts that were upregulated among homozygotes for c.1521_1523delCTT in CFTR, who carry two rare EHF alleles, were enriched for genes that alter protein glycosylation and trafficking, both mechanisms being pivotal for the effective targeting of fully functional p.Phe508del-CFTR to the apical membrane of epithelial cells. We conclude that EHF modifies the CF phenotype by altering capabilities of the epithelial cell to correctly process the folding and trafficking of mutant p.Phe508del-CFTR.
Similar content being viewed by others

Introduction
The autosomal-recessively inherited monogenic disease cystic fibrosis (CF; OMIM #219700)1 is caused by two defective copies of the cystic fibrosis transmembrane conductance regulator (CFTR) gene,2 which encodes a chloride3 and bicarbonate4, 5 channel localized in the apical membrane of epithelial cells. The CFTR-transmitted basic defect can be used to diagnose the disease by the analysis of sweat glands,6, 7 nasal6, 7 or intestinal epithelium.7
As CF is a rare disease affecting about 1:2000 newborns in the Caucasian population, sample sizes of several 10 000 individuals desirable for many genome-wide association studies (GWAS)8 cannot be obtained for the rare disease CF. To preserve reasonable power to detect CF-modifying genes,9 the European CF Twin and Sibling Study restricts the analysis to homozygotes of c.1521_1523delCTT in CFTR, studies extreme phenotypes10, 11 in a case–reference setting and employs endophenotypes12, 13 rather than global clinical variables such as lung function.
Recently, Wright et al have described and replicated a significant association signal on a 11p13 intergenic region.14 We wanted to know whether we could reproduce this finding in our truly independent patient cohort of homozygotes for c.1521_1523delCTT in CFTR from the European CF Twin and Sibling Study, which differs from the North American CF Genetic Modifier Study by recruitment strategy, phenotype evaluation, choice of genetic markers and approach to evaluate genetic data as outlined before.9 In addition, we have studied transcriptome data from rectal suction biopsies, as first, intestinal epithelium expresses large amounts of CFTR15 and second, owing to the high turnover rate of epithelial cells in the intestine, these samples are less prone to secondary alterations by inflammatory processes in comparison to pulmonary tissue.16
Patients and methods
Measurement of the CF basic defect
Assessment of the CF basic defect was carried out in vivo by nasal potential difference (NPD) measurement and ex vivo on rectal suction biopsies by intestinal current measurement (ICM). As outlined in detail elsewhere, secretagogues that activate or block ion channels, ion exchangers or components of the cellular signal transduction pathways were applied by superfusion of the lower nasal turbinate17 or to excised rectal suction biopsies mounted in a micro-Ussing chamber.18
We have used both techniques to discriminate between patients with and without residual chloride secretion. As ICM is an ex-vivo method applied to patient’s biopsies, the toxic compound DIDS (4,48-diisothiocyanostilbene-2,28-disulfonic acid), which has been reported to block chloride channels other than CFTR, could be used to differentiate between CFTR-mediated residual chloride secretion and chloride secretion through alternative channels.19 Based on compounds used in NPD, contrasting phenotypes for the response to the applied secretagogues were defined whereby the work reported upon here is focused on the response to amiloride, which blocks the epithelial sodium channel ENaC. All ICM and NPD results obtained from twins and siblings, which were used to characterize the role of EHF in CF, have been previously described by Bronsveld et al.19, 20
Patient population
The study population and the selection criteria for cases and references of the association study have been described in detail elsewhere.9 Briefly, genotyping data from 101 CF families, 85 of which are a subgroup of the twin and sibling study panel of 466 twin and sibling pairs, were used for the association study.9 Sixteen unrelated homozygotes for c.1521_1523delCTT in CFTR with known basic defect and their parents were included into the analysis of the manifestation of the CF basic defect.9, 21, 22, 23 For the endophenotype ‘response to amiloride in NPD’, patients who showed a change of 27 mV or less upon superfusion of the lower nasal turbinate with amiloride-containing solution were defined as cases (13 unrelated patients).9 Patients who showed a change of 28 mV or more upon superfusion of the lower nasal turbinate with amiloride-containing solution were defined as references (17 unrelated patients).9
For the endophenotype ‘CFTR-mediated residual chloride secretion in ICM’, patients who displayed chloride secretion mediated by CFTR, defined as the presence of chloride secretion upon stimulation with carbachol and the presence of chloride secretion upon stimulation with histamine after inhibition with DIDS,19, 20 were enrolled as cases (nine unrelated patients).9 Contrastingly, patients who did not show residual chloride secretion upon stimulation with carbachol and histamine were enrolled as references (14 unrelated patients).9
EHF genotyping
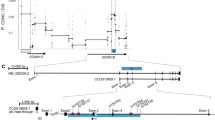
We have developed two microsatellite markers for EHF genotyping de novo as follows: the genomic sequence of the epithelial-specific transcription factor EHF (ets homologous factor, alias EHF, #26298; ENTREZ gene accessed at http://www.ncbi.nlm.nih.gov/gene) was obtained from NG_029177.1 (version from 02. APR 2012; bp coordinates 5001–47 248; ENTREZ nucleotide accessed at http://www.ncbi.nlm.nih.gov/nuccore), which covers the entire EHF-coding sequence. Four intragenic repetitive motifs were noticed: a (TG)n repeat in intron 1, two further (TG)n-repeats in intron 2 and a (CA)n-repeat in intron 6. Microsatellite genotyping using one biotinylated primer was established on a set of five non-CF control DNA samples. Two out of these four repetitive elements displayed allele variation among the control samples and were used to assess the genetic variability of the EHF locus. Microsatellite genotyping was established for two intragenic informative polymorphic sequences, that is, a (TG)n-repeat in intron 1 (motif starting at position 6071; primers used for amplification: 5′-TGTTGGGTCAGAGTGAATGG-3′ and 5′-ATCTCCCTGCTACCCACCTT-3′) and a (TG)n-repeat in intron 2 (motif starting at position 24 984; primers used for amplification: 5′-GGCAGTGGGATATCAGTCCA-3′ and 5′-GCTTATTGTCCATACCCAAATCG-3′ of the reference sequence (Figure 1a).

Allele distribution at the EHF locus. Markers EHFSat1 and EHFSat2 were genotyped on 101 CF families with a total of 171 patients who are homozygous for c.1521_1523delCTT in CFTR. Alleles at EHFSat1 and EHFSat2 were scored using an invariant set of control samples in arbitrary repeat units of the repetitive (TG)n-repeat motif. EHFsat1-EHFsat2 haplotypes of CF patients were reconstructed using FAMHAP. (a) Map of EHF, retrieved from http://www.ncbi.nlm.nih.gov/. EHFSat1 corresponds to the polymorphic sequence starting at position 6071; primers used for amplification: 5′-TGTTGGGTCAGAGTGAATGG-3′ and 5′-ATCTCCCTGCTACCCACCTT-3′. EHFSat2 corresponds to the polymorphic sequence starting at position 24 984; primers used for amplification: 5′-GGCAGTGGGATATCAGTCCA-3′ and 5′-GCTTATTGTCCATACCCAAATCG-3′; (b–d) Allele distribution for EHFSat1 (b), EHFSat2 (c) and marker combination EHFSat1-EHFSat2 (d) of the 171 homozygotes for c.1521_1523delCTT in CFTR from 101 CF families. (e and f) Allele distribution for marker combination EHFSat1-EHFSat2 among patient subsets stratified for response to amiloride in nasal potential difference measurement ((e): case population: 13 unrelated homozygotes for c.1521_1523delCTT in CFTR, which display a response of 27 mV or less to amiloride in NPD; reference population: 17 homozygotes for c.1521_1523delCTT in CFTR, which display a response of 28 mV or more to amiloride in NPD) and manifestation of DIDS-insensitive residual chloride secretion in intestinal current measurement ((f): case population: 9 unrelated homozygotes for c.1521_1523delCTT in CFTR, which display DIDS-insensitive residual chloride secretion in ICM; reference population: 14 unrelated homozygotes for c.1521_1523delCTT in CFTR, which do not display any residual chloride secretion in ICM).
Both markers were amplified using one biotinylated primer. Genotypes were visualized by direct blotting electrophoresis on a high-resolution polyacrylamide gel and chemoluminescence detection of biotinylated PCR products as described elsewhere.9 An invariant set of control samples was used as an internal control to calibrate the allele size on all genotyping analyses.
Data evaluation and statistics
Genotyping data were evaluated using the software package FAMHAP (http://famhap.meb.uni-bonn.de/). Genetic data for the association study were evaluated using the FAMHAP software package,24 which allows family-based analysis 25, 26 and accepts data evaluation in association with studies on unrelated individuals as well as on affected sib pairs.24 Case–reference comparisons were carried out using 10 000 Monte–Carlo simulated data sets,24, 25, 26 whereby the analysis of more than one marker per locus is corrected for multiple testing by haplotype permutation.26 For this purpose, the entire data set of cases and references is used to estimate haplotype frequencies.24 Haplotype, or, in cases of non-informative phase or haplotype uncertainty, weighted haplotype explanation lists are assigned to each individual whereby the haplotype frequencies of the entire data set are taken into account to compute the conditional likelihood weights.24 Permutation is done by randomly assigning the affection status to the individuals in each replication whereby the ratio of cases to controls is kept constant.24
P-values for comparison of n-marker-haplotypes and all marker subsets derived thereof are computed as s/n, where n is the number of permutation replicates and s is the number of permutation replicates leading to a test statistic higher than or equal to that of the real data.24 Reported P values are: Praw, referring to a computed P value of a single marker or marker combination and Pcorr, referring to the P value of the entire marker set that is corrected for multiple testing of all genotyped markers. The adjustment for multiple testing properly accounts for LD within the Monte–Carlo simulation framework that evaluates the corrected significance minP, the smallest observed raw P-value.25 The computational details of the minP principle have been described elsewhere.25
To allow a comparable assignment of weighted haplotype explanations in all subpopulations, the entire genotyping data of 101 CF families were provided as a training set to FAMHAP for all case–reference comparisons.9
Transcriptome analysis
The global transcriptome data evaluated within this manuscript were derived from an earlier study on rectal suction biopsies, which include 16 unrelated homozygotes for c.1521_1523delCTT in CFTR, who were enrolled for their ICM phenotype as described above.21, 22, 23 Explicitly, the basic defect and subsequently the transcriptome were assessed in the same patients and in the same tissue samples.
Total RNA was isolated from rectal suction biopsies using the RNeasy kit (Qiagen Corp, Hilden, Germany) and quality was determined by gel electrophoresis. cDNA was synthesized from total RNA and hybridized on the Affymetrix Chips (Gene-ChipHuman Genome U133 Plus 2.0 Array, Affymetrix, Santa Clara, CA, USA) according to the protocol from Affymetrix Manual (version 700217 rev 3). The data set has been deposited in the GEO database under accession number GSE15568. The expression data for 22283 probe sets were normalized and evaluated using Affymetrix Microarray Suite v5.1 software (Affymetrix).
The transcriptiome data were analyzed using the web-based interface of the tool GenePattern, accessed at http://broadinstitute.org/cancer/software/genepattern,27 to evaluate our transcriptome data comparing two patient subgroups defined by their EHF intragenic background at 22 283 probe sets. Next, we have submitted the two lists of differentially expressed genes to the database for annotation, visualization and integrated discovery DAVID, accessed at http://david.abcc.ncifcrf.gov/,28, 29 for gene-enrichment analysis.
Results
Allele distribution at the EHF locus
We have developed two microsatellite markers EHFSat1 and EHFSat2 within EHF. Both were informative showing a bimodal allele distribution, displaying a polymorphism information content of 0.26 (EHFSat1) and 0.37 (EHFSat1) and heterozygosity values30 of 0.51 (EHFSat1) and 0.73 (EHFSat2), respectively (Figure 1a–c).
Haplotype frequencies derived by family-based analysis through FAMHAP24, 25, 26 for marker combination EHFSat1-EHFSat2 showed no divergence from expectancy values assuming an independent combination of alleles at both marker loci, indicating that variants at both markers have arisen sufficiently long ago to reflect a low LD (D′=0.172) between the two polymorphic markers, which are located at a physical distance of 18 kb. EHFSat1-EHFSat2 haplotype 10-14 was observed on a third of the chromosomes and three further haplotypes (10-10, 10-13, 15-14) were observed on a tenth of chromosomes. The remaining 50% of chromosomes carried 1 of the 15 other rare EHFSat1-EHFSat2 haplotypes (Figure 1d).
Minor indication of a transmission disequilibrium (Praw=0.0712) that did not withstand the correction for multiple testing (Pcorr=0.168) was observed whereby EHF haplotypes 10-10 and 10-13 were being more frequently observed on transmitted than on non-transmitted alleles, whereas the haplotype 10-14 was enriched among non-transmitted alleles.
In conclusion, the two markers EHFSat1 and EHFSat2 were sensitive with respect to differences in the EHF genetic background.
Association of EHF alleles with the manifestation of the CF basic defect
We wanted to know whether the EHF genetic background was associated with the manifestation of the CF basic defect. Case and reference populations were defined based on NPD measurements and ICM. 17, 18
Significant differences between case and reference populations have been observed for the manifestation of the CF basic defect in respiratory and intestinal tissue (Figure 1e). First, allele distributions for marker combination EHFSat1-EHFSat2 were different between patients with high (n=17) and low (n=13) response to amiloride in NPD measurement (Praw=0.0082, Pcorr=0.02). Second, allele distributions for marker combination EHFSat1-EHFSat2 were different between patients with DIDS-insensitive residual chloride secretion (n=9) and patients who did not show any residual chloride secretion (n=14) as determined by ICM (Praw=0.0268, Pcorr=0.0593).
The allele distributions of the patient subpopulation who displayed a high response to amiloride in NPD and the patient subpopulation who did not show any residual chloride secretion in ICM were both markedly enriched for the most frequent EHF allele designated 10-14 at marker combination EHFSat1-EHFSat2. In both case subpopulations, nobody carried this most frequent EHF allele on both chromosomes, whereas in both reference subpopulations, four of the patients were homozygous for the most frequent EHF allele 10-14 (endophenotype NPD/amiloride response: Praw=0.0474 for diplotype analysis; endophenotype ICM/DIDS-insensitive residual chloride conductance: Praw=0.0531 for diplotype analysis).
We conclude that the most frequent EHF allele, designated as 10-14 in our marker combination EHFSat1-EHFSat2, was associated with a CF-typical basic defect phenotype that is characterized by a high response to amiloride in NPD and by the absence of residual chloride secretion in ICM. Conversely, rare EHF alleles were associated with DIDS-insensitive residual chloride secretion, which is indicative of CFTR-mediated residual activity in the homozygotes for c.1521_1523delCTT in CFTR.
EHF genetic background defines a set of differentially expressed genes
We wanted to know how the genetic background of EHF, encoding for an epithelial-specific transcription factor,31, 32, 33 alters the transcriptome of epithelial cells in order to provide a molecular hypothesis for the observed association of rare EHF alleles with CFTR-mediated residual chloride secretion. We have reviewed global transcriptome data from 16 unrelated homozygotes for c.1521_1523delCTT in CFTR who have been enrolled in a study on the manifestation of the basic defect in excised intestinal biopsies and subsequent CFTR protein analysis and GeneChip-based transcriptome analysis.9, 21, 22, 23
The 16 homozygotes for c.1521_1523delCTT in CFTR enrolled into the transcriptome study were grouped based on their intragenic EHF haplotypes. As the most frequent EHF haplotype was associated with a CF-typical basic defect phenotype, that is, high response to amiloride and the absence of CFTR-mediated residual function, we decided to define two subgroups, one of which carries two rare EHF alleles (seven patients) and one of which carries at least one frequent EHF allele (nine patients).
We have used GenePattern27 to evaluate our transcriptome data comparing two patient subgroups defined by their EHF intragenic background at 22 283 probe sets provided by hybridization of cDNA derived from rectal suction biopsies of homozygous patients for c.1521_1523delCTT in CFTR to Gene-ChipHuman Genome U133 Plus 2.0 Array (Affymetrix,Santa Clara, CA, USA). Using the formal non-stringent cutoff of P<0.05 to select differentially expressed genes between our two subsamples stratified for EHF genotype, we could identify 1166 probe sets as candidates (Supplementary material, Supplementary Table 1). Expression values of 746 of these probe sets were elevated among the nine homozygotes for c.1521_1523delCTT in CFTR, which carry at least one frequent EHF allele, and expression levels of 420 probe sets were elevated among the seven homozygotes for c.1521_1523delCTT in CFTR, which carry two rare EHF alleles.
Genes that define protein trafficking and processing are upregulated among carriers of rare EHF alleles
We have submitted the two lists of differentially expressed genes to the database for annotation, visualization and integrated discovery DAVID28, 29 for gene enrichment analysis. For the analysis of the data presented here, gene sets defined by a common annotation were considered based on the Gene Ontology—categories34 for ‘cellular component’ (GOTERM_CC), ‘molecular function’ (GOTERM_MF) and ‘biological process’ (GOTERM_BP).
The primary DAVID output was filtered using a stringent EASE–score28, 29 of 0.001 and demanding that the retrieved GO gene set is covered by more than 10 genes within the analyzed data set, equivalent to a coverage of at least 2.6% (11/420) and 1.5% (11/746) of the differentially expressed genes. By these criteria, the two data sets were enriched for gene sets with entirely different functions: The data set of the 746 probe sets with elevated expression values among the nine homozygotes for c.1521_1523delCTT in CFTR, which carry at least one frequent EHF allele, genes that interact with DNA and/or RNA, being likely to participate in general gene regulation, is enriched (Supplementary Table 3). In contrast, in the 420 probe sets with higher expression among the seven homozygotes for c.1521_1523delCTT in CFTR, which carry two rare EHF alleles, genes that specifically take part in the glycosylation of biopolymers and genes that are assigned to the Golgi apparatus were found to be enriched by DAVID (Supplementary Table 4). As p.Phe508del-CFTR is well-known as a folding and trafficking mutant,1, 35 genes that alter membrane protein glycolsylation and/or transport within the Golgi apparatus such as those found among enriched among these 420 genes are likely candidates to mediate the observed association between a rare EHF background and CFTR-mediated residual chloride secretion.
EHF differentially expressed genes encode for CFTR interaction partners
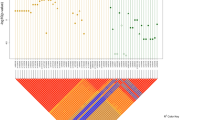
We wanted to know how many of the 1166 differentially expressed genes can be associated with protein processing, trafficking and post-translational modification or protein degradation via any of the above-listed CFTR-relevant pathways in order to provide a second mode of data analysis independent of the hypothesis-free gene-enrichment analysis. As DAVID could provide an annotation for 378 out of the 1166 genes submitted to the annotation tools with our stringent cutoff criteria of an cutoff of EASE score of 0.001 and a gene count of more than 10 genes, we decided to screen the entire data set of 1166 differentially expressed genes manually for annotations containing terms related to trafficking, processing of membrane proteins, vesicle trafficking and subcellular designations to the compartments ‘endoplasmatic reticulum (ER)’, ‘golgi’, ‘endocytic vesicle’, ‘lysosome’ and ‘proteasome’. A total of 98 gene records could be attributed to one of these categories or were associated with vesicle transport, listed as members of the heat-shock protein family directly interacting with CFTR (HSPA4, alias Hsp70) or designated as part of the ubiquitin-dependent degradation pathway (Figure 2). Please note that the distribution of genes is highly asymmetrical between the two subgroups defined by EHF intragenic backgound: genes associated with proteasomal degradation (Figure 2i) are predominatly upregulated among carriers of the frequent EHF haplotype. Most strikingly, genes associated with post-translational modifications (Figure 2e) are nearly exclusively found in the gene set that is upregulated among carriers of rare EHF haplotypes.

EHF-dependent gene regulation in relation to p.Phe508del-CFTR biosynthesis, trafficking and post-translational modification. Mature, fully glycosylated and functional p.Phe508del-CFTR—emphasized as a green line at the apical membrane (AM) – reaches the apical membrane through trafficking pathways36, 37 illustrated as green arrows (a). These encompass biosynthesis and insertion into the lipid bilayer (IA), utilization of the ER-associated folding pathway (ERAF)37, passage through the ER, ERGIC (IB) and Golgi-compartments, post-translational modifications (PTMs) and finally, transport of mature p.Phe508del-CFTR to subapically localized vesicles (IC) and to the AM (ID). The alternative CFTR maturation pathway that bypasses the ERGIC and Golgi compartments49 is shown (II). In contrast, pathways that lead to degradation of p.Phe508del-CFTR36, 37 are depicted in orange. These encompass the ER-associated degradation pathway ER-associated degradation (ERAD)37 that leads to degradation in the proteasome (PR) and the retrograde traffic of endosomes from the subapical compartment (III) toward the lysosome (LY). EHF-dependent differentially regulated genes whose products have been annotated to partake in any of these pathways crucial to p.Phe508del-CFTR biosynthesis, maturation and trafficking are shown in b–j. Gene products for which the subcellular localization cannot be specified are listed in j. Forty trafficking and maturation genes whose expression are upregulated among the nine p.Phe508del-CFTR homozygous carriers of at least one frequent EHF allele are shown in orange. Fifty-eight trafficking and maturation genes whose expression are upregulated among the seven p.Phe508del-CFTR homozygous carriers of two rare EHF alleles are shown in green.
A detailed description of the so-called chaperome, describing CFTR interaction partners has been provided by Wang et al.37 Curiously, among the 1166 differentially expressed probe sets, the following 12 genes are part of the CFTR chaperome: the sorting nexin SNX4, the culumenin CALU and the kinesin KIF5B (involved in protein sorting and/or vesicle transport), the proteasome subunit PSMB1, the cystatin B CSTB and the ubiquitin C (involved in protein degradation), the cytoplasmatic guanine nucleotide exchange factor RANBP10, the GTPase SEPT6, the ATPase ATP2A2, the regulatory protein FAM120A, the apoptosis-inducing factor AIFM1 and the regulatory membrane-protein of complement-mediated cell lysis CD59. Related findings are: the sorting nexin 13, the gene EPS8L1, whereby the epidermal growth factor receptor pathway substrate 8, EPS8, is part of the p.Phe508del-CFTR chaperome,37 and the cadherin CDH11, whereby CDH1, is part of the p.Phe508del-CFTR chaperome.37 In conclusion, part of the chaperome that determines whether or not p.Phe508del-CFTR is processed correctly, appears to be regulated by an EHF-dependent pathway. Apart from these genes allocated to the CFTR chaperome, the CFTR interaction partner syntaxin 1638 and the chaperone HSPA4 (heat shock 70kDa protein 4) are among the differentially regulated genes.
EHF differentially expressed genes encode for CF modifiers
Of the 1166 differentially expressed probe sets, four CF-modifying genes and/or their networking partners have been implicated. Of the tumor necrosis factor receptor 1 pathway, TNFR1 itself being a CF modifier,10 the genes TNFAIP2 (tumor necrosis factor, alpha-induced protein 2) and TRAF4 (TNF receptor-associated factor 4) have been found to be differentially regulated. The signal transducer and activator of transcription 3 STAT3 were identified as a CF modifier26 and as a differentially regulated gene itself as well as PIAS4 (protein inhibitor of activated STAT). The CF modifier ADRB2,8 encoding the beta2-adrenergic receptor, was likewise present among the EHF-regulated genes. Finally, the network of the CF modifier transforming growth factor beta 18 was represented by four genes: TGIF2 (TGFB-induced factor homeobox 2), LTBP1 (latent transforming growth factor beta-binding protein 1), LTBP2 (latent transforming growth factor beta-binding protein 2) and TGFBI (transforming growth factor, beta-induced). Notably, STAT3 and TNFR1 have been shown to modify the CF basic defect.10, 26 In conclusion, gene products belonging to pathways of CF-modifying genes are represented among the data set of EHF-dependent, differentially expressed genes. Finally, an inhibitor of the histone deacetylase 7 was shown to restore the function of p.Phe508del-CFTR.39 Consistently, histone deacetylase 7 was 2.3-fold downregulated among the p.Phe508del-CFTR homozygotes, which carry two rare EHF alleles associated with CFTR-mediated residual function in our sample.
Discussion
In 2010, Wright et al have described a CF modifier locus on 11p13 in a GWAS,15 suggesting the epithelial-specific transcription factor EHF18, 19, 20 as a strong candidate. We have asked whether we could reproduce the finding and elucidate the underlying molecular mechanism and have undertaken a replication study in the patient panel of homozygotes for c.1521_1523delCTT in CFTR of the European CF Twin and Sibling Study.
To compensate for the loss of power inflicted upon the study by the limited sample size of subgroups stratified for extreme clinical phenotypes (being infrequent by definition) or for basic defect phenotypes, such as homozygotes for c.1521_1523delCTT in CFTR, which display CFTR-mediated residual function (being infrequent because of the etiology of the disease), care was taken to use highly informative markers to target EHF as a candidate gene in our study population.
Association signals were observed for two basic defect manifestations of perturbed ion transport: first, the manifestation of a CF-typical high response to amiloride in NPD measurement was associated with the most frequently observed EHF allele observed on a third of the chromosomes (Praw=0.0082, Pcorr=0.02). Second, rare EHF alleles were accumulated among patients with CFTR-mediated residual chloride secretion determined by ICM of rectal suction biopsies (Praw=0.0268, Pcorr=0.0593). These two independent findings in non-overlapping subsamples both assign the most frequent EHF allele to the CF-typical phenotype, whereas in the both cases, rare EHF alleles correspond to atypical basic defect phenotypes among homozygotes for c.1521_1523delCTT in CFTR.
To investigate how rare and frequent EHF alleles translate on the molecular level, we have reviewed transcriptome data obtained from rectal suction biopsies with Affymetrix gene chips.10 Data sets were compared between samples from individuals who carry two rare EHF alleles and samples from individuals with at least one frequent EHF allele. Comparison of transcriptomes by GenePattern31 and pathway analysis by DAVID 32, 33 indicated that the transcript set upregulated among carriers of rare EHF alleles was significantly enriched for genes that participate in post-translational modification of proteins by glycosylation and proteins that facilitate trafficking between the ER and Golgi compartment.
The CF disease causing lesion c.1521_1523delCTT in CFTR has been termed a folding and trafficking mutant as the efficiency with which the resulting mutant protein p.Phe508del-CFTR is transported to the apical membrane and processed to the fully glycosylated form is substantially lower in comparison to wild-type CFTR protein.36 The amount of correctly processed and functional p.Phe508del-CFTR protein is increased by efficient CFTR gene transcription, processing of the primary transcript by splicing and subsequent biosynthesis of the membrane protein in the ER. Subsequently, efficient trafficking requires correct folding of transmembrane helices through the ER lipid bilayer,40, 41 passage through the endoplasmic reticulum–Golgi intermediate compartment (ERGIC) and Golgi compartments,42, 43, 44 glycosylation and finally translocation to the apical membrane.45 Analogously, the amount of apically located CFTR is decreased by retention of the protein in the ER and subsequent degradation by the proteasome through the ER-associated degradationpathway.42, 43, 44 Part of the cellular CFTR pool resides in a subapical membrane compartment from which it can be either recycled to the membrane or destined for degradation.46, 47, 48 An alternative trafficking pathway that bypasses the ERGIC and Golgi-compartments has been described by Yoo et al.49 Strikingly, genes involved in these pathways that are vital for the processing and trafficking of membrane proteins such as CFTR were differentially expressed in intestinal epithelial tissue derived from homozygotes for c.1521_1523delCTT in CFTR who were stratified according to their EHF genetic background (Figure 2).
In conclusion, the data presented in this work shows consistency with the current knowledge on p.Phe508del-CFTR cellular physiology as an alteration of two major pathways, that is, protein glycosylation and trafficking, promotes p.Phe508del-CFTR residual function depending on the EHF genetic background. As the amount of residual chloride secretion has been shown to correlate with CF disease severity before,24, 50, 51 it is tempting to speculate that the association signal on 11p13, detected in a GWAS conducted to identify modifiers for CF lung disease severity in the large patient cohort of the North American CF Genetic Modifier Study,15 has its molecular basis in the EHF-dependent alteration of the epithelial transcriptome.
Accession codes
References
Welsh MJ, Ramsey BW, Accurso F, Cutting GR : Cystic Fibrosis, in Scriver CR (ed). The Metabolic & Molecular Bases of Inherited Disease: New York, NY, 2001, pp 5121–5188.
Strausbaugh SD, Davis PB : Cystic fibrosis: a review of epidemiology and pathobiology. Clin Chest Med 2007; 28: 279–288.
Rich DP, Anderson MP, Gregory RJ et al: Expression of cystic fibrosis transmembrane conductance regulator corrects defective chloride channel regulation in cystic fibrosis airway epithelial cells. Nature 1990; 347: 358–363.
Tang L, Fatehi M, Linsdell P : Mechanism of direct bicarbonate transport by the CFTR anion channel. J Cyst Fibros 2009; 8: 115–121.
Paradiso AM, Coakley RD, Boucher RC : Polarized distribution of HCO3- transport in human normal and cystic fibrosis nasal epithelia. J Physiol 2003; 548: 203–218.
Farrell PM, Rosenstein BJ, White TB et al: Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J Pediatr 2008; 153: S4–S14.
De Boeck K, Derichs N, Fajac I et al: New clinical diagnostic procedures for cystic fibrosis in Europe. J Cyst Fibros 2011; 10 (Suppl 2): S53–S66.
Collaco JM, Cutting GR : Environmental and non-CFTR modifiers of cystic fibrosis. CML—Cystic Fibrosis 2012; 2: 57–65.
Spencer CC, Su Z, Donnelly P, Marchini J : Designing genome-wide association studies: sample size, power, imputation, and the choice of genotyping chip. PLoS Genet 2009; 5: e1000477.
Stanke F, Becker T, Kumar V et al: Genes that determine immunology and inflammation modify the basic defect of impaired ion conductance in cystic fibrosis epithelia. J Med Genet 2011; 48: 24–31.
Risch N, Zhang H : Extreme discordant sib pairs for mapping quantitative trait loci in humans. Science 1995; 268: 1584–1589.
Jacquemont S, Reymond A, Zufferey F et al: Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature 2011; 478: 97–102.
Gottesman II, Gould TD: The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry 2003; 160: 636–645.
Rommelse NN, Arias-Vasquez A, Altink ME et al: Neuropsychological endophenotype approach to genome-wide linkage analysis identifies susceptibility loci for ADHD on 2q21.1 and 13q12.11. Am J Hum Genet 2008; 83: 99–105.
Wright FA, Strug LJ, Doshi VK et al: Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat Genet 2011; 43: 539–546.
Strong TV, Boehm K, Collins FS : Localization of cystic fibrosis transmembrane conductance regulator mRNA in the human gastrointestinal tract by in situ hybridization. J Clin Invest 1994; 93: 347–354.
Meyerholz DK, Stoltz DA, Pezzulo AA, Welsh MJ : Pathology of gastrointestinal organs in a porcine model of cystic fibrosis. Am J Pathol 2010; 176: 1377–1389.
Bochert MA, Kleinbaum LA, Sun LY, Burton FH : Molecular cloning and expression of Ehf, a new member of the ets transcription factor/oncoprotein gene family. Biochem Biophys Res Commun 1998; 246: 176–181.
Kas K, Finger E, Grall F et al: ESE-3, a novel member of an epithelium-specific ets transcription factor subfamily, demonstrates different target gene specificity from ESE-1. J Biol Chem 2000; 275: 2986–2998.
Kleinbaum LA, Duggan C, Ferreira E, Coffey GP, Buttice G, Burton FH : Human chromosomal localization, tissue/tumor expression, and regulatory function of the ets family gene EHF. Biochem Biophys Res Commun 1999; 264: 119–126.
Schüler D, Sermet-Gaudelus I, Wilschanski M et al: Basic protocol for transepithelial nasal potential difference measurements. J Cyst Fibros 2004; 3 (Suppl 2): 151–155.
De Jonge HR, Ballmann M, Veeze H et al: Ex vivo CF diagnosis by intestinal current measurements (ICM) in small aperture, circulating Ussing chambers. J Cyst Fibros 2004; 3 (Suppl 2): 159–163.
Bronsveld I, Mekus F, Bijman J et al: Residual chloride secretion in intestinal tissue of deltaF508 homozygous twins and siblings with cystic fibrosis. The European CF Twin and Sibling Study Consortium. Gastroenterology 2000; 119: 32–40.
Bronsveld I, Mekus F, Bijman J et al: Chloride conductance and genetic background modulate the cystic fibrosis phenotype of Delta F508 homozygous twins and siblings. J Clin Invest 2001; 108: 1705–1715.
Kumar V, Becker T, Jansen S et al: Expression levels of FAS are regulated through an evolutionary conserved element in intron 2, which modulates cystic fibrosis disease severity. Genes Immun 2008; 9: 689–696.
Labenski H, Hedtfeld S, Becker T, Tümmler B, Stanke F : Initial interrogation, confirmation and fine mapping of modifying genes: STAT3, IL1B and IFNGR1 determine cystic fibrosis disease manifestation. Eur J Hum Genet 2011; 19: 1281–1288.
van Barneveld A, Stanke F, Tamm S et al: Functional analysis of F508del CFTR in native human colon. Biochim Biophys Acta 2010; 1802: 1062–1069.
Herold C, Becker T : Genetic association analysis with FAMHAP: a major program update. Bioinformatics 2009; 25: 134–136.
Knapp M, Becker T : Family-based association analysis with tightly linked markers. Hum Hered 2003; 56: 2–9.
Becker T, Knapp M : A powerful strategy to account for multiple testing in the context of haplotype analysis. Am J Hum Genet 2004; 75: 561–570.
Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP : GenePattern 2.0. Nat Genet 2006; 38: 500–501.
Huang DW, Sherman BT, Lempicki RA : Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4: 44–57.
Huang DW, Sherman BT, Lempicki RA : Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37: 1–13.
Shete S, Tiwari H, Elston RC : On estimating the heterozygosity and polymorphism information content value. Theor Popul Biol 2000; 57: 265–271.
Ashburner M, Ball CA, Blake JA et al: Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25: 25–29.
Lukacs GL, Verkman AS : CFTR: Folding, misfolding and correcting the ΔF508 conformational defect. Trends Mol Med 2012; 18: 81–91.
Wang X, Venable J, LaPointe P et al: Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 2006; 127: 803–815.
Gee HY, Tang BL, Kim KH, Lee MG : Syntaxin 16 binds to cystic fibrosis transmembrane conductance regulator and regulates its membrane trafficking in epithelial cells. J Biol Chem 2010; 285: 35519–35527.
Hutt DM, Herman D, Rodrigues AP et al: Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat Chem Biol 2010; 6: 25–33.
Cui L, Aleksandrov L, Chang XB et al: Domain interdependence in the biosynthetic assembly of CFTR. J Mol Biol 2007; 365: 981–994.
Chen EY, Bartlett MC, Loo TW, Clarke DM : The DeltaF508 mutation disrupts packing of the transmembrane segments of the cystic fibrosis transmembrane conductance regulator. J Biol Chem 2004; 279: 39620–39627.
Wang X, Koulov AV, Kellner WA, Riordan JR, Balch WE : Chemical and biological folding contribute to temperature-sensitive DeltaF508 CFTR trafficking. Traffic 2008; 9: 1878–1893.
Wang X, Matteson J, An Y et al: COPII-dependent export of cystic fibrosis transmembrane conductance regulator from the ER uses a di-acidic exit code. J Cell Biol 2004; 167: 65–74.
Bannykh SI, Bannykh GI, Fish KN, Moyer BD, Riordan JR, Balch WE : Traffic pattern of cystic fibrosis transmembrane regulator through the early exocytic pathway. Traffic 2000; 1: 852–870.
Gentzsch M, Chang XB, Cui L et al: Endocytic trafficking routes of wild type and DeltaF508 cystic fibrosis transmembrane conductance regulator. Mol Biol Cell 2004; 15: 2684–2696.
Swiatecka-Urban A, Duhaime M, Coutermarsh B et al: PDZ domain interaction controls the endocytic recycling of the cystic fibrosis transmembrane conductance regulator. J Biol Chem 2002; 277: 40099–40105.
Swiatecka-Urban A, Brown A, Moreau-Marquis S et al: The short apical membrane half-life of rescued {Delta}F508-cystic fibrosis transmembrane conductance regulator (CFTR) results from accelerated endocytosis of {Delta}F508-CFTR in polarized human airway epithelial cells. J Biol Chem 2005; 280: 36762–36772.
Ameen N, Silvis M, Bradbury NA : Endocytic trafficking of CFTR in health and disease. J Cyst Fibros 2007; 6: 1–14.
Yoo JS, Moyer BD, Bannykh S, Yoo HM, Riordan JR, Balch WE : Non-conventional trafficking of the cystic fibrosis transmembrane conductance regulator through the early secretory pathway. J Biol Chem 2002; 277: 11401–11409.
Dray-Charier N, Paul A, Scoazec JY et al: Expression of delta F508 cystic fibrosis transmembrane conductance regulator protein and related chloride transport properties in the gallbladder epithelium from cystic fibrosis patients. Hepatology 1999; 29: 1624–1634.
Sermet-Gaudelus I, Vallee B, Urbin I et al: Normal function of the cystic fibrosis conductance regulator protein can be associated with homozygous (Delta)F508 mutation. Pediatr Res 2002; 52: 628–635.
Acknowledgements
We acknowledge funding from the Deutsche Forschungsgemeinschaft (SFB 621, project C7), the European Union (QL G1-CF- 2001-01005), the BMBF (German Center for Lung Research, partner site BREATH, Disease Area CF), the Fritz-Thyssen-Stiftung (Az.20.08.0.044), the Deutsche Fördergesellschaft für die Mukoviszidoseforschung e.V. and the Mukoviszidose e.V.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
SW, AvB, FS worked on the evaluation of transcriptome data; SH worked on the EHF genotyping; TB worked on the EHF genotype data analysis. BT and FS designed the project and wrote the manuscript.
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Stanke, F., van Barneveld, A., Hedtfeld, S. et al. The CF-modifying gene EHF promotes p.Phe508del-CFTR residual function by altering protein glycosylation and trafficking in epithelial cells. Eur J Hum Genet 22, 660–666 (2014). https://doi.org/10.1038/ejhg.2013.209
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.209
Keywords
This article is cited by
-
Interstitielle Prozesse der Lunge im Kindesalter
Der Pathologe (2017)
-
Deep resequencing of CFTR in 762 F508del homozygotes reveals clusters of non-coding variants associated with cystic fibrosis disease traits
Human Genome Variation (2016)
-
Pluripotent stem cells for disease modeling and drug screening: new perspectives for treatment of cystic fibrosis?
Molecular and Cellular Pediatrics (2015)
-
Genome-wide association meta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis
Nature Communications (2015)
