
Abstract
Genome-wide association studies (GWAS) have identified numerous prostate cancer-associated risk loci. Some variants at these loci may be regulatory and influence expression of nearby genes. Such loci are known as cis-expression quantitative trait loci (cis-eQTL). As cis-eQTLs are highly tissue-specific, we asked if GWAS-identified prostate cancer risk loci are cis-eQTLs in human prostate tumor tissues. We investigated 50 prostate cancer samples for their genotype at 59 prostate cancer risk-associated single-nucleotide polymorphisms (SNPs) and performed cis-eQTL analysis of transcripts from paired primary tumors within two megabase windows. We tested 586 transcript–genotype associations, of which 27 were significant (false discovery rate ≤10%). An equivalent eQTL analysis of the same prostate cancer risk loci in lymphoblastoid cell lines did not result in any significant associations. The top-ranked cis-eQTL involved the IRX4 (Iroquois homeobox protein 4) transcript and rs12653946, tagged by rs10866528 in our study (P=4.91 × 10−5). Replication studies, linkage disequilibrium, and imputation analyses highlight population specificity at this locus. We independently validated IRX4 as a potential prostate cancer risk gene through cis-eQTL analysis of prostate cancer risk variants. Cis-eQTL analysis in relevant tissues, even with a small sample size, can be a powerful method to expedite functional follow-up of GWAS.
Similar content being viewed by others

Introduction
Genome-wide association studies (GWAS) in prostate cancer (PrCa) have identified numerous replicable risk loci.1, 2, 3, 4, 5, 6, 7, 8, 9, 10 One of the current major challenges is to understand the underlying biology behind these association signals. Unlike most genetic effectors of Mendelian diseases, the majority of GWAS-discovered risk variants tend to lie within intronic, intergenic, or gene desert regions.
One hypothesis is that these risk loci may contain variants located within regulatory elements, therefore modulating expression levels of nearby genes.11 Gene expression levels are known to be heritable.12, 13, 14 Genetic variants correlated with nearby transcript levels are defined as cis-expression quantitative trait loci (cis-eQTL). Indeed, GWAS-ascertained phenotype-associated single-nucleotide polymorphisms (SNPs) are enriched in cis-eQTLs.15
It is recognized that cis-eQTLs are highly tissue-specific.16, 17 However, the acquisition of the appropriate tissue type is logistically challenging and the majority of eQTL studies were based on lymphoblastoid cell lines (LCLs). Recently, Grisanzio et al18 reported five cis-eQTLs in prostate tissue (both normal and tumor) that alter expression of nearby genes.18 Here, we present an unbiased cis-eQTL analysis of PrCa GWAS variants in prostate tumor tissue.
Materials and methods
eQTL study subjects and tissue samples
Prostate samples were collected from 50 patients of European ancestry with localized or locally advanced PrCa treated with radical prostatectomy as monotherapy, as described previously.19 The samples were collected as part of an IRB-approved protocol at Weill Cornell Medical College (New York, NY, USA).
RNA-seq and genotype data for cis-eQTL analysis
Total RNA was extracted with TRIzol (Invitrogen, Carlsbad, CA, USA) from frozen tissue blocks selected for high-density cancer foci and then prepared for sequencing with Illumina’s (San Diego, CA, USA) sample preparation protocol for paired-end (PE) sequencing of mRNA, with slight modifications as described previously.19 In brief, 5–10 μg of total RNA was heat-fragmented, reverse-transcribed with Superscript II (Invitrogen), and size-selected on a gel before ligation of the PE adapters. PE RNA-seq was performed on the Genome Analyzer II (Illumina) and reads were aligned using ELAND to the human genome (hg18/NCBI36). The average number of mapped reads per sample was around ten million. Mapped transcript levels were quantified in terms of RPKM (Reads Per Kilobase per Million) using RSEQTools.20 The log 2 of RPKMs plus one was evaluated and the data were then quantile-normalized.
Genotype data from Affymetrix SNP 6.0 (Santa Clara, CA, USA) array data was generated from normal tissue as described previously.21, 22 Only SNPs for which genotypes could be called for, at least 95% of the samples and for which the probe sequence specificity was compliant with empirically determined parameters,23 were utilized.
SNP selection
SNPs used in the cis-eQTL analysis were selected as follows: 51 SNPs associated with PrCa from the NHGRI Catalog (accessed February 2011),24 one SNP associated with PrCa from a fine mapping study of the 10q11 risk locus25 and seven SNPs associated with levels of prostate secretions.26, 27 For each SNP not present on the Affymetrix 6.0 array, pairwise linkage disequilibrium (LD), as measured by r2, was computed using genotype data from residents of Utah with northern and western European ancestry (HapMap3 CEU population; Altshuler et al;28 http://hapmap.ncbi.nlm.nih.gov/). For each SNP, the SNP on the Affymetrix 6.0 array with the strongest LD was selected as the tag SNP. For comprehensiveness, we included poorly tagging SNPs to insure that any strong eQTLs at those loci would be discovered, even at the cost of increasing the number of tests and potentially missing some weaker eQTLs after correcting for multiple testing. Three SNPs failed quality control filters as described previously.23 Supplementry Table S1 lists all SNPs tested.
Cis-eQTL statistical analysis
Cis-eQTL analysis was performed as described previously.29 Briefly, we performed cis analysis by first identifying all genes whose transcriptional midpoint are within 1 Mb of the SNP being tested. We then tested each SNP–transcript pair for an allele dosage effect of SNP on expression levels in a linear model. To account for multiple hypothesis testing, we defined statistical significance as having a false discovery rate (FDR) <10%.30
LCL eQTL analysis
Microarray data previously generated on the Illumina WG-6 microarray platform31 on 60 CEU EBV-transformed LCLs was correlated with genotypes generated by the HapMap project.32 Statistical analysis was the same as described above for the prostate tissue eQTL analysis.
Copy number change analysis at the IRX4 locus
To investigate the presence of somatic copy number changes at the IRX4 (Iroquois homeobox protein 4) locus, Affymetrix 6.0 data from the corresponding tumor tissues were queried. Tumor data were preprocessed as described previously21 and data normalization was performed against the paired normal samples to correct for germline copy number variants.
Imputation and fine mapping association
PrCa case–control sets of European Americans (1235 cases, 1214 controls) from the Cancer Genetic Markers of Susceptibility (CGEMS) study33 and Japanese Americans (1040 cases, 1093 controls) and African Americans (2327 cases, 2508 controls) from the GENEVA study of the Multiethnic Cohort34 were obtained from dbGaP (accessions phs000207.v.1.p1 and phs000306.v.2.p1, respectively). The data sets used for the analyses described in this manuscript were obtained from dbGaP at https://dbgap.ncbi.nlm.nih.gov. Additional SNPs in the 50 kilobase (kb) region flanking rs10866528 (chr5: 1 866 772–1 916 869, hg19/GRCh37) were imputed using IMPUTE 2.1.2 (ref. 35) with the 1000 Genomes October 2011 (ref. 36) data set as reference. Association analyses with the imputed data set were performed in SNPTEST under an additive model.37
Association analysis in an Ashkenazi Jewish PrCa case and control study
Ashkenanzi Jewish prostate case and control samples were obtained under IRB-approved protocols as described previously.38 Briefly, PrCa cases presented at Memorial Sloan-Kettering Cancer Center (MSKCC) with histologically confirmed PrCa. Control DNA came from healthy men in New York. All cases and controls have self-reported four grandparents of Ashkenanzi ancestry.
Samples were genotyped with a Taqman SNP Genotyping Assay (Applied Biosystems, Carlsbad, CA, USA) (C_1186265_10 for rs12653946) in an ABI 7900HT according to the manufacturer’s directions. Association analyses were performed in PLINK v.1.07 under an allelic model.39 The genotyping rate for rs12653946 was 90%. Rs12653946 did not deviate from Hardy–Weinberg equilibrium in this study population (P=0.9193).
Association in the Tyrol Prostate-Specific Antigen Screening Cohort
The Tyrol Prostate-Specific Antigen Screening Cohort was recently genotyped for a case–control study on inherited copy number variants.40 SNP genotype information for rs10866528 was interrogated for association with PrCa risk.
Results
Association between PrCa risk loci and expression levels of nearby genes
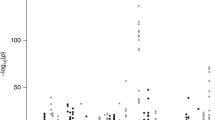
Fifty-nine SNPs (tagging 52 PrCa risk SNPs and 7 SNPs associated with prostate secretions) spanning 39 distinct PrCa risk loci were tested for association with expression levels of nearby transcripts (Supplementry Table S1). Out of 586 total SNP–transcript associations tested, 27 had an FDR of <10% (Supplementry Table S2). Removal of poorly tagged SNPs (r2 in CEU <0.7) and redundant eQTLs due to strong LD between tested SNPs resulted in 14 high-quality putative eQTLs spread over seven distinct loci (Table 1). We tested the tissue specificity of these putative eQTLs by performing an equivalent analysis in a set of 60 LCLs. Out of the same list of 52 PrCa risk SNPs, none were found to be eQTLs in LCLs with an FDR <10%.
The strongest association was between IRX4 and rs10866528 (P=4.91 × 10−5, FDR=0.00468). The genotype associated with the highest risk of PrCa was correlated with lower IRX4 expression (Figure 1). Rs10866528 tags the PrCa risk SNP rs12653946, a risk SNP originally discovered in a Japanese population.5 The two SNPs are in perfect LD in HapMap’s Japanese in Tokyo, Japan (JPT) cohort. There was no evidence of somatic copy number changes at this locus in our prostate tumor samples. The remaining statistically significant eQTLs were considerably weaker associations (P=0.0141–0.002) (Table 1).

An expression quantitative trait locus for the transcription factor IRX4 RNA expression levels (mapped on a log(RPKM+1) scale) of the IRX4 transcript by genotype is plotted as a boxplot, with the median as the middle bar and the ends of the box representing the 25th and 75th percentiles. RPKM=Reads Per Kilobase per Million mapped reads.
Population-dependent association at the IRX4 locus
As the association between SNPs near IRX4 and PrCa was first reported in a Japanese population,5 despite previous larger GWAS in populations of European descent, we hypothesized that there might be population differences in the effect of this SNP on PrCa risk. A literature review of the original GWAS and all subsequent replication studies of the association of rs12653946 with PrCa risk revealed a population effect on the strength of association (Table 2). The odds ratio (OR) of rs12653946’s association with PrCa in the original Japanese study was similar to subsequent replication studies in the Han Chinese population (OR=1.26–1.41),41, 42 whereas studies in populations of European descent found lower ORs overall (OR=1.11–1.20).43, 44, 45 In contrast, a replication study carried out in African Americans did not report a statistically significant association (OR=1.05).46 The minor allele at rs12653946 is common across the studied populations with a minor allele frequency (MAF) of 0.35–0.44 and is also uniformly the risk allele.
One possibility that may explain this discrepancy among populations is that the true causative variant may be better tagged in Asian populations by rs12653946 than in other populations studied. Indeed, differences in LD structure in HapMap3 populations reveal divergence at this locus (Figure 2).

Population-specific PrCa association results after imputation at 5p15. Association results are overlaid over colored representations of r2 from rs12653946 (purple diamond), as visualized with LocusZoom.53
The correlation between rs10866528 and rs12653946 genotype also differs by ethnicity (Supplementry Table S3). In the HapMap3 Japanese (JPT) samples, the two SNPs are perfectly correlated with r2=1. In Chinese from Beijing, China (CHB) and Chinese from Denver, Colorado (CHD), LD is still strong, with r2 of 0.972 and 0.951, respectively. LD begins to lower in CEU with r2=0.849, and drops drastically in Yorubans from Nigeria (YRI), with r2=0.659.
Imputation of 5p15 in multiple populations
To account for fine-scale LD structure at this locus and to determine if variants with higher effect sizes could be detected in other populations, we imputed a 50 kb genomic region flanking rs10866528 in three different ethnic populations (Japanese-American, European-American and African-American), with data sets available in NIH’s Database of Genotypes and Phenotypes (dbGaP). After imputation using reference panels available from the 1000 Genomes Project, we performed an association analysis to ask which variants were most strongly associated with PrCa in each population (Figure 2). Our analysis revealed a block of strong LD of about 7 kb in the Japanese population that included rs10866528 and rs12653946. This block was relatively uniform in its strong association with PrCa. There were no study-wide significant associations found in any of the other populations.
Association of IRX4 SNPs with PrCa in Ashkenanzi Jewish and European case–control studies
We also tested whether SNPs at the IRX4 locus were associated with PrCa in two case–control studies of PrCa risk (Table 3). In a study of Ashkenanzi Jewish PrCa cases and controls in New York,38 we found evidence for association of rs12653946 with OR=1.18 (95% CI: 1.02–1.37) at a P-value of 0.0286. We were unable to test the association of rs10866528 with PrCa in our Ashkenanzi Jewish group because of technical problems with the stock TaqMan assay. No association was found between rs10866528 and PrCa in the Tyrol Prostate-Specific Antigen Screening Cohort (P=0.9892; OR=1.002).40 We did not test rs12653946 in this cohort as our analysis was based on existing Affymetrix SNP 6.0 microarray data.
Discussion
Our cis-eQTL study based on 50 prostate tumor samples found evidence for several eQTLs at PrCa risk loci. The strongest eQTL’s gene target, IRX4, was not known to have a role in PrCa at the initiation of the study. IRX4 is a transcription factor that has previously been studied in the context of vertebrate heart chamber patterning.47 An IRX4-deficient mouse had normal embryonic cardiac morphology and adult-onset cardiomyopathy.48 Recently, a targeted cis-eQTL study in Japanese PrCa tissues reported the rs12653946-IRX4 eQTL.49 In concordance with our study, they observed that the PrCa risk allele at rs12653946 correlated with lower expression of the IRX4 transcript in prostate tissue. Their functional analyses found several variants upstream of IRX4 that alter transcriptional activity, as well as a tumor-suppressive role for IRX4 in PrCa cells. Our analyses extend the finding of a cis-eQTL at this locus to a European population.
We found no evidence for somatic copy number alterations at IRX4 in the prostate tumors studied, suggesting that, at least in our study, such somatic alterations do not contribute to variability in IRX4 expression.
Our exploration of the IRX4 locus in several populations revealed a population specificity for the strength of association of rs12653946 with PrCa; studies in Japanese and Han Chinese populations displayed a stronger association than in non-Asian populations. Several explanations may account for this difference. Fine-scale LD is variable at this locus and there may be population-specific tagging of the causative variant by the risk SNP. As we were not able to find a strong association signal in European-American or African-American individuals after imputing existing GWAS data, we doubt that such population-specific tagging of a common variant explains the population differences. Although gene–environment interaction is a formal possibility, our results argue against it. Japanese Americans in Hawaii and Los Angeles display the same strong association as Japanese in Tokyo, whereas no association is observed in African Americans from Los Angeles. Alternatively, the causative variant may be a rare variant of large effect and may be even less common in non-Asian versus Asian populations or different causal rare variants or gene–gene interaction effects may exist in different populations at this locus. We note that the magnitude of effect of the SNP on IRX4 expression levels appears similar in our study of individuals of European ancestry and previously reported data from Japanese individuals,49 raising the possibility that the population differences of the SNP effect on disease risk are due to factor(s) acting downstream of IRX4.
Although the correlation between rs10866528 and rs12653946 also weakened from Asian populations to European and African-American populations, the causative variant for PrCa risk does not appear to be better tagged by rs10866528, as there is clearly no evidence for association between rs10866528 and PrCa in a Tyrol-based European population.
Among the weaker associations discovered in our study, several are likely to be true positives. An eQTL in prostate tissues at 10q11 for NCOA4, a known androgen receptor coactivator, has been reported previously.50 Rs5945619 has been also found to be an eQTL for NUDT11,18 a diphosphoinositol polyphosphate phosphohydrolase. Lastly, rs7250689 has been previously found to be associated with levels of PPP1R14A in monocytes (eQTL browser, http://eqtl.uchicago.edu/).51 In summary, out of the seven distinct high-quality, statistically significant eQTLs discovered in our study, four have been previously described as eQTLs, and three in prostate normal and tumor tissue specifically. Out of the gene targets for the three eQTLs that had not been described previously, FAM83F has no known protein function; FOXP4 is a transcription factor involved in development; and CYBRD1 is a reductase enzyme involved in iron metabolism in the duodenum. None of these have any reported connection to PrCa.
Simultaneously, we found no evidence for eQTLs at any of the known PrCa risk loci in LCLs, suggesting that the putative eQTLs are tissue specific. We note that as we only considered potential eQTLs for which the FDR was below 10%, there could be less significant eQTLs for PrCa risk SNPs observable in LCLs. However, even if this were the case, it would not discount from the observation that many PrCa risk SNPs are eQTLs in prostate tumors at an FDR of 10%, whereas no such associations are observed in LCLs with a comparable sample size. Although many tissues can contribute to disease predisposition, this observation emphasizes the importance of considering tissue type carefully before undertaking an eQTL study with known disease risk SNPs. We note that in this study, we only considered prostate tumors. Other tissue types, such as benign prostate tissue and stromal tissue, may also have a role in PrCa etiology. Therefore, although such tissue types were not available for this study, a comprehensive eQTL study of PrCa risk loci should include those tissues as well.
We did not replicate a known PrCa eQTL for rs10993994 at microseminoprotein-β (MSMB). We had previously observed that rs10993994 and other SNPs at 10q11 are associated with levels of β-MSP, the protein product of MSMB.26 Others had observed association between rs10993994 and MSMB mRNA levels in both prostate tumor and normal tissue, although the signal was stronger in normal tissue.50 Although we did not observe a study-wide statistically significant correlation of SNPs at 10q11 with MSMB levels, we note that rs3123078 at 10q11 was nominally associated with MSMB levels (P=0.047). Rs3123078 is a PrCa risk SNP10 and resides 24.6 kb upstream of MSMB. We hypothesize that several factors combined to result in only observing such a weak association. The sample size of this study was modest (n=50), limiting our power. Moreover, the strongest risk SNP at MSMB, rs10993994, was only tagged at r2=0.7 in this study, meaning our effective sample size for testing rs10993994 was n=35. As we examined prostate tumors rather than normal tissues, the expected effect size for this association was also attenuated, further reducing power.50
Similarly, no statistically significant eQTL was observed at the other 32 distinct PrCa loci tested. Besides the aforementioned possible reasons, there may be biological reasons for a lack of correlation with cis-gene expression. The causal risk variant may be regulatory through a different mechanism, such as alternative splicing. The causal risk variant may regulate expression in a different tissue or temporal context. For instance, PrCa risk SNPs at the 8q24 locus appear to regulate c-myc expression patterns, but these same risk SNPs are not correlated with steady-state c-myc expression levels.52 The risk locus may be due to a non-regulatory mechanism, such as tagging for a coding variant. Although more comprehensive and larger eQTL studies in appropriate tissues are warranted, such analyses alone are unlikely to resolve fully the question of how prostate associated risk loci contribute to disease development.
In summary, we have provided evidence for seven eQTLs among known PrCa risk SNPs. Although cis-eQTL analysis is a study of correlation and not causation, it is a powerful and efficient way to generate biological hypotheses about the functional mechanism of common risk variants.
References
Murabito JM, Rosenberg CL, Finger D et al: A genome-wide association study of breast and prostate cancer in the NHLBI's Framingham Heart Study. BMC Med Genet 2007; 8 ((Suppl 1):): S6.
Gudmundsson J, Sulem P, Steinthorsdottir V et al: Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat Genet 2007; 39: 977–983.
Gudmundsson J, Sulem P, Manolescu A et al: Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet 2007; 39: 631–637.
Gudmundsson J, Sulem P, Rafnar T et al: Common sequence variants on 2p15 and Xp11.22 confer susceptibility to prostate cancer. Nat Genet 2008; 40: 281–283.
Takata R, Akamatsu S, Kubo M et al: Genome-wide association study identifies five new susceptibility loci for prostate cancer in the Japanese population. Nat Genet 2010; 42: 751–754.
Gudmundsson J, Sulem P, Gudbjartsson DF et al: Genome-wide association and replication studies identify four variants associated with prostate cancer susceptibility. Nat Genet 2009; 41: 1122–1126.
Eeles RA, Kote-Jarai Z, Giles GG et al: Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet 2008; 40: 316–321.
Thomas G, Jacobs KB, Yeager M et al: Multiple loci identified in a genome-wide association study of prostate cancer. NatgGenet 2008; 40: 310–315.
Sun J, Zheng SL, Wiklund F et al: Sequence variants at 22q13 are associated with prostate cancer risk. Cancer Res 2009; 69: 10–15.
Eeles RA, Kote-Jarai Z, Al Olama AA et al: Identification of seven new prostate cancer susceptibility loci through a genome-wide association study. Nat Genet 2009; 41: 1116–1121.
Maurano MT, Humbert R, Rynes E et al: Systematic localization of common disease-associated variation in regulatory DNA. Science (New York, NY) 2012; 337: 1190–1195.
Morley M, Molony CM, Weber TM et al: Genetic analysis of genome-wide variation in human gene expression. Nature 2004; 430: 743–747.
Cheung VG, Conlin LK, Weber TM et al: Natural variation in human gene expression assessed in lymphoblastoid cells. Nat Genet 2003; 33: 422–425.
Schadt EE, Monks SA, Drake TA et al: Genetics of gene expression surveyed in maize, mouse and man. Nature 2003; 422: 297–302.
Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ : Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet 2010; 6: e1000888.
Dimas AS, Deutsch S, Stranger BE et al: Common regulatory variation impacts gene expression in a cell type-dependent manner. Science (New York, NY) 2009; 325: 1246–1250.
Nica AC, Parts L, Glass D et al: The Architecture of Gene Regulatory Variation across Multiple Human Tissues: The MuTHER Study. PLoS Genet 2011; 7: e1002003.
Grisanzio C, Werner L, Takeda D et al: Genetic and functional analyses implicate the NUDT11, HNF1B, and SLC22A3 genes in prostate cancer pathogenesis. Proc Natl Acad Sci USA 2012; 109: 11252–11257.
Pflueger D, Terry S, Sboner A et al: Discovery of non-ETS gene fusions in human prostate cancer using next-generation RNA sequencing. Genome Res 2011; 21: 56–67.
Habegger L, Sboner A, Gianoulis TA et al: RSEQtools: a modular framework to analyze RNA-Seq data using compact, anonymized data summaries. Bioinformatics 2011; 27: 281–283.
Barbieri CE, Baca SC, Lawrence MS et al: Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet 2012; 44: 685–689.
Setlur SR, Chen CX, Hossain RR et al: Genetic variation of genes involved in dihydrotestosterone metabolism and the risk of prostate cancer. Cancer Epidemiol Biomarkers Prev 2010; 19: 229–239.
Oldridge DA, Banerjee S, Setlur SR, Sboner A, Demichelis F : Optimizing copy number variation analysis using genome-wide short sequence oligonucleotide arrays. Nucleic Acids Res 2010; 38: 3275–3286.
Hindorff LA, Sethupathy P, Junkins HA et al: Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA 2009; 106: 9362–9367.
Chang B-L, Cramer SD, Wiklund F et al: Fine mapping association study and functional analysis implicate a SNP in MSMB at 10q11 as a causal variant for prostate cancer risk. Hum Mol Genet 2009; 18: 1368–1375.
Xu X, Valtonen-André C, Sävblom C, Halldén C, Lilja H, Klein RJ : Polymorphisms at the Microseminoprotein-beta locus associated with physiologic variation in beta-microseminoprotein and prostate-specific antigen levels. Cancer Epidemiol Biomarkers Prev 2010; 19: 2035–2042.
Gudmundsson J, Besenbacher S, Sulem P et al: Genetic correction of PSA values using sequence variants associated with PSA levels. Sci Transl Med 2010; 2: 62ra92.
Altshuler DM, Gibbs RA, Peltonen L et alInternational HapMap 3 Consortium: Integrating common and rare genetic variation in diverse human populations. Nature 2010; 467: 52–58.
Banerjee S, Oldridge D, Poptsova M, Hussain WM, Chakravarty D, Demichelis F : A computational framework discovers new copy number variants with functional importance. PLoS One 2011; 6: e17539.
Benjamini Y, Hochberg Y : Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 1995; 57: 289–300.
Stranger BE, Nica AC, Forrest MS et al: Population genomics of human gene expression. Nat Genet 2007; 39: 1217–1224.
Frazer KA, Ballinger DG, Cox DR et alInternational HapMap Consortium: A second generation human haplotype map of over 3.1 million SNPs. Nature 2007; 449: 851–861.
Yeager M, Orr N, Hayes RB et al: Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet 2007; 39: 645–649.
Haiman CA, Patterson N, Freedman ML et al: Multiple regions within 8q24 independently affect risk for prostate cancer. Nat Genet 2007; 39: 638–644.
Howie BN, Donnelly P, Marchini J : A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 2009; 5: e1000529.
1000 Genomes Project Consortium: A map of human genome variation from population-scale sequencing. Nature 2010; 467: 1061–1073.
Marchini J, Howie B : Genotype imputation for genome-wide association studies. Nat Rev Genet 2010; 11: 499–511.
Vijai J, Kirchhoff T, Gallagher D et al: Genetic architecture of prostate cancer in the Ashkenazi Jewish population. Br J Cancer 2011; 105: 864–869.
Purcell S, Neale B, Todd-Brown K et al: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Demichelis F, Setlur SR, Banerjee S et al: Identification of functionally active, low frequency copy number variants at 15q21.3 and 12q21.31 associated with prostate cancer risk. Proc Natl Acad Sci USA 2012; 109: 6686–6691.
Long Q-Z, Du Y-F, Ding X-Y et al: Replication and fine mapping for association of the C2orf43, FOXP4, GPRC6A and RFX6 genes with prostate cancer in the Chinese population. PLoS One 2012; 7: e37866.
Wang M, Liu F, Hsing AW et al: Replication and cumulative effects of GWAS-identified genetic variations for prostate cancer in Asians: a case–control study in the ChinaPCa consortium. Carcinogenesis 2012; 33: 356–360.
Batra J, Lose F, Chambers S et al: A replication study examining novel common single nucleotide polymorphisms identified through a prostate cancer genome-wide association study in a Japanese population. Am J Epidemiol 2011; 174: 1391–1395.
Schumacher FR, Berndt SI, Siddiq A et al: Genome-wide association study identifies new prostate cancer susceptibility loci. Hum Mol Genet 2011; 20: 3867–3875.
Lindstrom S, Schumacher FR, Campa D et al: Replication of five prostate cancer loci identified in an Asian population – results from the NCI Breast and Prostate Cancer Cohort Consortium (BPC3). Cancer Epidemiol Biomarkers Prev 2012; 21: 212–216.
Haiman CA, Chen GK, Blot WJ et al: Characterizing genetic risk at known prostate cancer susceptibility loci in African Americans. PLoS Genet 2011; 7: e1001387.
Bao ZZ, Bruneau BG, Seidman JG, Seidman CE, Cepko CL : Regulation of chamber-specific gene expression in the developing heart by Irx4. Science (New York, NY) 1999; 283: 1161–1164.
Bruneau BG, Bao ZZ, Fatkin D et al: Cardiomyopathy in Irx4-deficient mice is preceded by abnormal ventricular gene expression. Mol Cell Biol 2001; 21: 1730–1736.
Nguyen HH, Takata R, Akamatsu S et al: IRX4 at 5p15 suppresses prostate cancer growth through the interaction with vitamin D receptor, conferring prostate cancer susceptibility. HumMol Genet 2012; 21: 2076–2085.
Pomerantz MM, Shrestha Y, Flavin RJ et al: Analysis of the 10q11 cancer risk locus implicates MSMB and NCOA4 in human prostate tumorigenesis. PLoS Genet 2010; 6: e1001204.
Zeller T, Wild P, Szymczak S et al: Genetics and beyond – the transcriptome of human monocytes and disease susceptibility. PLoS One 2010; 5: e10693.
Grisanzio C, Freedman ML : Chromosome 8q24-associated cancers and MYC. Genes Cancer 2010; 1: 555–559.
Pruim RJ, Welch RP, Sanna S et al: LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics (Oxford, UK) 2010; 26: 2336–2337.
Acknowledgements
This research was supported by a grant from the Geoffrey Beene Cancer Research Center (RJK). JV was supported by the Niehaus Clinical Genetics Initiative and the Sharon Levine Corzine Fund at MSKCC. FD was supported by Associazione Italiana Ricerca Cancro (AIRC). We are grateful to the study investigators and funding agencies that supported the work deposited in dbGaP used in this study. Funding support for the CGEMS study of prostate cancer, and the epidemiological studies that provided the samples for the GWAS, was provided by NIH Grants CA CA55075, 5U01CA098233-04 U01 CA098710 and NIH Contracts N01-CN-45165, N01-RC-45035 and N01-RC-37004. Funding support for the GENEVA Prostate Cancer study was provided through the National Cancer Institute (R37CA54281, R01CA63464, P01CA33619, U01CA136792, U01CA98758 and RC2 CA148085) and the National Human Genome Research Institute (U01HG004726). Assistance with phenotype harmonization, SNP selection, data cleaning, meta-analyses, data management and dissemination, and general study coordination was provided by the GENEVA Coordinating Center (U01HG004789-01). We also thank Naoki Kitabayashi for technical support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Xu, X., Hussain, W., Vijai, J. et al. Variants at IRX4 as prostate cancer expression quantitative trait loci. Eur J Hum Genet 22, 558–563 (2014). https://doi.org/10.1038/ejhg.2013.195
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.195
Keywords
This article is cited by
-
A biallelic multiple nucleotide length polymorphism explains functional causality at 5p15.33 prostate cancer risk locus
Nature Communications (2023)
-
Gene regulatory mechanisms underpinning prostate cancer susceptibility
Nature Genetics (2016)
-
ASEQ: fast allele-specific studies from next-generation sequencing data
BMC Medical Genomics (2015)
