
Abstract
Dystroglycanopathies are a genetically heterogeneous subset of congenital muscular dystrophies that exhibit autosomal recessive inheritance and are characterized by abnormal glycosylation of α-dystroglycan. In particular, POMT2 (protein O-mannosyltransferase-2) mutations have been identified in congenital muscular dystrophy patients with a wide range of clinical involvement, ranging from the severe muscle-eye-brain disease and Walker–Warburg syndrome to limb girdle muscular dystrophy without structural brain or ocular involvement. Cardiovascular disease is thought to be uncommon in congenital muscular dystrophy, with rare reports of cardiac involvement. We describe three brothers aged 21, 19, and 17 years with an apparently homozygous POMT2 mutation who all presented with congenital muscular dystrophy, intellectual disabilities, and distinct cardiac abnormalities. All three brothers were homozygous for a p.Tyr666Cys missense mutation in exon 19 of the POMT2 gene. On screening echocardiograms, all siblings demonstrated significant dilatation of the aortic root and depressed left ventricular systolic function and/or left ventricular wall motion abnormalities. Our report is the first to document an association between POMT2 mutations and aortopathy with concomitant depressed left ventricular systolic function. On the basis of our findings, we suggest patients with POMT2 gene mutations be screened not only for myocardial dysfunction but also for aortopathy. In addition, given the potential for progression of myocardial dysfunction and/or aortic dilatation, longitudinal surveillance imaging is recommended both for patients with disease as well as those that have normal baseline imaging.
Similar content being viewed by others


Introduction
Dystroglycanopathies are a subgroup of CMDs with autosomal recessive inheritance.1 They may result from abnormal glycosylation of α-dystroglycan.2 Although CMDs may exhibit only congenital hypotonia, the most severe clinical features in CMD may include muscle, eye, and/or brain pathology. This triad is found in muscle-eye-brain disease (MEB; MIM 253280) and Fukuyama congenital muscular dystrophy (FCMD; MIM 253800) that together with Walker–Warburg syndrome (WWS; MIM 236670) include the disorders termed cobblestone lissencephalies.3 WWS is a congenital condition characterized by the hydrocephalus, agyria, and retinal dysplasia with or without encephalocele.
Individuals with α-dystroglycanopathies have been shown to have mutations in the genes POMT1, POMT2, FKRP, KFNT, POMGnT1, DPM3, and LARGE.3 Protein O-mannosyltransferase 2 (POMT2) is a 21-exon gene that encodes an integral membrane protein of the endoplasmic reticulum required for the correct glycosylation of α- dystroglycan.4, 5 Both POMT1 and POMT2 mutations are the most common causes of WWS.1, 2, 6, 7
Cardiac involvement among the dystroglycanopathies remains poorly characterized. Whereas dilated and hypertrophic cardiomyopathy have been described,8 aortopathy in dystroglycanopathies has not been previously reported.
We describe three brothers who harbor a mutation in the POMT2 gene in association with dilatation of the annulus, sinotubular junction, aortic root, and ascending aorta with concomitant depressed left ventricular (LV) systolic function.
Patients, Methods and Results
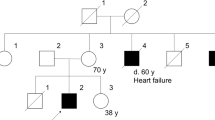
We describe three siblings in descending chronological age. Specific details about their past medical and family history were unavailable because of family and social issues prohibiting evaluation of the mother or any other biological relatives.
Patient 1initially presented at the age of 17 years. He was one of three full siblings, all of whom were referred for evaluation of suspected myopathy of unknown type. He had a history of cognitive impairment, congenital hypotonia, and generalized muscle weakness. He additionally had neurofibromatosis type 1. There was no history of seizures. He was in special education classes in the tenth grade. He was able to walk for short distances but preferred to use a manual or power wheelchair for mobility most of the day.
He was under Child Protective Services (CPS) custody and living in a group home. Details of the history include the pregnancy, perinatal and early-childhood course were limited. However, the father was known to be deceased. He had neurofibromatosis and reportedly died of cancer. Two half-sisters through the father also had neurofibromatosis but no reported history of muscle disease. The mother’s medical history was unknown.
Physical examination revealed normal vital signs without need for respiratory support. He had small body habitus with height and weight less <5 percentile. His head circumference was below the second percentile (51.5 cm) with mild dolichocephaly. He had a large area of hyperpigmentation on the right side of his face. There were multiple café-au-lait spots on all of his extremities and trunk along with axillary freckling. There were several small molluscum fibrosum over his trunk. He had a 60° right thoracic scoliosis from T3 to T12 and a 36° left thoracolumbar curve from T12 to L5. There were flexion contractures in the hips, elbows, and knees.
Neurological examination demonstrated good expressive articulation, although he had echolalia. Cranial nerve evaluation was normal, including normal facial strength, full extraocular movements, and no tongue atrophy. There were no cataracts or other eye abnormalities. There was decreased muscle bulk in the proximal and distal extremities. Strength examination demonstrated proximal weakness in the shoulder girdle and hips (MRC 4-/5), whereas his distal power was preserved (MRC 5/5). He had a Gower sign in arising from the floor but was able to independently ambulate with accentuation of his lumbar lordosis in a Trendelenberg gait. Reflexes were absent throughout.
Laboratory results showed an elevated creatine kinase of 4335 U/l (upper limits or normal 370 U/l). Dystrophin mutation analysis, including deletion, duplication, and DNA sequencing, was negative for mutations. A vastus lateralis muscle biopsy demonstrated findings consistent with a dystrophic myopathy. Immunostaining for dystrophin showed positive staining with normal quantity of dystrophin with the rod domain antibody. The C-term antibody showed positive staining with no significant variation in staining between and within fibers. Immunostaining for alpha, beta and gamma sarcoglycans showed no significant reduction in the staining pattern. Immunofluorescence for merosin revealed slightly reduced staining. Immunofluorescence for dysferlin showed mild cytoplasmic accumulation and slightly variable membrane expression on many fibers. Caveolin 3 was present and shows slight variation in staining. Alpha dystroglycan staining was markedly reduced with variable mosaic staining. The pattern of staining for beta dystroglycan and beta spectrin controls was similar. (Figures 1 and 2) Emerin staining was positive. There was no up-regulation of expression of utrophin. Immunoblots showed normal quantity of dystrophin protein with appropriate size with the rod antibody. The C-term antibody showed normal quantity of dystrophin protein with appropriate size of protein. Immunoblots for alpha sarcoglycan and beta dystroglycan showed slight reduction of expression. Alpha dystroglycan was reduced with absence of the 156 kDa protein band. Calpain 3 was present with reduced protein. The dysferlin protein was detectable with normal sized protein but with reduced quantity of protein.

Immunofluorescence with antigen specific antibodies show the following: (a) normal staining for alpha dystroglycan in a control; (b) lack of staining for alpha dystroglycan (patient); (c) markedly reduced staining for Dysferlin; (d) moderately reduced staining for Merosin; (e) normal staining for beta dystroglycan and markedly reduced staining for (f) alpha sarcoglycan, (g) beta sarcoglycan, and (h) gamma sarcoglycan.

(a) H&E stain showing moderate variation in fiber size, fiber splits, increased internalized nuclei and mild increase in the endomysial connective tissue. Inset shows regenerating fibers. (b) Modified Gomori trichrome stain showing a whorled hypertrophic fiber. Inset shows focal phagocytosis. Magnification × 200.
Subsequent mutation analysis of dystroglycan-related muscular dystrophy genes LARGE1, FKTN, FKRP, POMT1, POMT2, and POMGnT1 was performed and identified a known disease-causing homozygous missense mutation (c.1997A>G ) in exon 19 of POMT2.
Initial cardiovascular examination was at 17 years of age. His cardiac examination revealed a physiologically split second heart sound without murmur or gallop. An echocardiogram showed a normal annulus diameter and a dilated aortic root with a diameter of 2.94 cm (Z-score 3.49). The sinotubular junction (STJ) diameter was 2.37 cm (Z-score 2.91) and the ascending aortic diameter was 2.35 cm (Z-score 2.52). (Figure 3) The echocardiogram also demonstrated normal left ventricular size with a shortening fraction of 38% (normal value 30 to 42%) and some degree of dyskinesis in the inferior interventricular septum and the LV posterior wall. In response to the cardiovascular findings, medical therapy was instituted with a beta blocker (Atenolol 12.5 mg twice daily).

Patient 1 in a parasternal long-axis view shows his dilated aortic root from the initial cardiac evaluation. LA, left atrium; LV, left ventricle; RV, right ventricle.
A subsequent echocardiogram was performed at the age of 18 years that revealed a persistently dilated aortic root, STJ, and ascending aorta. In addition, the LV end diastolic dimension (LVEDD) measured 4.68 cm (Z-score 2.2) with an LV shortening fraction of 30%. Both, the septum and LV posterior wall continued to be mildly dyskinetic. Because of the changes in the LV function, an ACE inhibitor (Enalapril 2.5 mg twice daily) was added to his medical regimen.
Despite medical intervention, the most recent cardiology visit at the age of 21 years revealed a mildly dilated aortic annulus 1.99 cm (Z-score 2.43), a persistently dilated aortic root 2.71 cm (Z-score 2.43), and STJ segment with 2.32 cm (Z-score 2.5). Although the ascending aorta was not able to be visualized, an improvement in the LV systolic function was observed, with a shortening fraction of 36.5% possibly secondary to the institution of medical therapy.
Patient 2 presented at 15 years of age along with his older brother described above. He also had a history of cognitive impairment, congenital hypotonia, and weakness along with epilepsy, which was well controlled with lamotrigine. He was not known to have neurofibromatosis. A previous muscle biopsy at the age of 6 years was reported to have shown findings consistent with a muscular dystrophy.
On physical examination, he had a small body habitus with height, weight, and head circumference less than the 5 percentile. His vital signs were unremarkable, and he did not require respiratory assistance. He had dolichocephaly. There were no clinical stigmata of neurofibromatosis. There were no cataracts or other eye abnormalities. His cranial nerves were intact without appreciable facial weakness. Strength examination found proximal weakness in the shoulder and hips (MRC 4/5). There were mild flexion contractures in the shoulders, elbows, finger flexors, hips, and knees. He had a Gower sign. He was able to walk short distances independently with a Trendeleberg gait. His reflexes were absent throughout. There was no Babinski sign.
His laboratory results were remarkable for a creatine kinase value of 5180 U/l. His first echocardiogram revealed a normal aortic annulus diameter measuring 1.60 cm (Z-score 0.56), moderate dilatation of the aortic root with a diameter of 2.90 cm (Z-score 4.17), and a normal STJ diameter measuring 1.90 cm (Z-score 1.13). The LV was mildly dilated, with an LVEDD of 4.60 cm (Z-score 2.15). The LV diastolic wall thickness was 0.50 cm (Z-score −2.61), with a calculated LV ejection fraction (LVEF) of 49% (mildly decreased). Given the depressed LV systolic function, the patient was started on enalapril 2.5 mg twice daily, which was tolerated without difficulty.
An MRI of the brain was performed at 19 years of age. This demonstrated low normal thickness of the corpus callosum on the sagittal T1-weighted midline imaging and prominence of the ventricular system suggestive of possible reduction in white matter volume. (Figure 4) Normal signal was returned from the deep white matter of both hemispheres. There was mild cerebellar volume loss with the prominence of median sulci, cerebellar folia, and ex vacuo dilatation of the fourth greater than bodies of the lateral ventricles (Figure 3).

An MRI of the brain from the second sibling shows borderline microcephaly, paucity of deep white matter, mild diffuse cerebellar and cerebral volume loss with ex vacuo dilatation of the fourth greater than bodies of the lateral ventricle.
Sequence analysis of POMT2 identified the same homozygous missense mutation in exon 19 as seen in his older brother.
In the most recent cardiac evaluation, his echocardiogram showed a mild progression of the enlarged aortic dimensions when compared with findings from the previous examination. The aortic root diameter measured 3.10 cm (Z-score 4.63), the STJ diameter was 2.50 cm (Z-score 3.68), and the ascending aorta diameter was 2.5 cm (Z-score 2.80) (Table 1). The LVEF remained mildly depressed with a calculated LVEF of 47% with normalization of his LVEDD. His Enalapril was increased to 5 mg twice daily without side effect. The addition of beta-blockade was deferred, as his blood pressures were in the lower range, and there was clinical concern that he would not tolerate both the increase in enalapril and addition of an additional blood pressure lowering therapy by his caregivers.
Patient 3 was initially evaluated at the age of 12 years and was noted to have cognitive impairment and proximal muscle weakness with gait failure at 10 years of age CMD. He used a power wheelchair for mobility, but he did not require respiratory assistance at the time of his initial evaluation. He had small body habitus like his brothers with height, weight, and head circumference less than the 5 percentile. He had dolichocephaly. There were no clinical stigmata of neurofibromatosis. Cranial nerves were intact. He had proximal weakness with flexion contractures in his shoulder, elbows, hips, knees, and ankles. Deep tendon reflexes were absent throughout.
His laboratory results were remarkable for a creatine kinase of 5700 U/l. Genetic analysis detected the same missense mutation seen in his brothers. An echocardiogram revealed dilatation of the aortic root measuring 2.90 cm (Z-score 3.50) and STJ measuring 2.30 cm (Z-score 2.71) with mild increase in the LVEDD measuring 5.70 cm (Z-score 4.09) (Figure 5). The calculated LVEF was 66%. Enalapril was instituted at 2.5 mg twice daily on the basis of findings of LV dilatation. At age sixteen, his aortic diameters demonstrated persistent enlargement. However, there was worsening of LV systolic function with a reduction of the LVEF to 44%. His Enalapril was increased to 5 mg twice daily, and a beta blocker (carvedilol) was started at 3.125 mg twice daily. Both of these interventions were tolerated without difficulty. During his last cardiology evaluation, the aortic root and STJ remained dilated but his LVEF improved to 50%. Ambulatory 24 h ECG monitoring was performed for arrhythmia surveillance in each brother and did not detect any significant rhythm disturbances or conduction system disease.

Patient 3 in a parasternal long-axis view from the most recent visit shows persistent dilatation of the aortic root and STJ. LA, left atrium; LV, left ventricle.
A summary of the clinical phenotypes is provided in Table 1.
Discussion
In this report, we describe three siblings who harbor a homozygous POMT2 mutation in association with dilatation of the aortic root and left ventricular dilatation, LV wall motion abnormalities and LV systolic dysfunction. The diagnosis of an α-dystroglycanopathy was based on the following: (1) the neurologic findings, (2) imaging results, (3) muscle biopsy, and (4) molecular analysis of the POMT2 gene.
Altered α-dystroglycan glycosylation has been associated with mutations in six genes (POMT1, POMT2, POMGnT1, fukutin, FKRP, and LARGE). O-mannosyl transferases 1 and 2 (POMT1, POMT2) are required for the attachment of O-linked tetrasaccharides to α-dystroglycan. O-mannose beta-1,2-N-acetylglucosaminyltransferase (POMGnT1) is required for the transfer of N-acetylglucosamine (GlcNAc) to mannose O-linked to α-dystroglycan. Fukutin, fukutin-related protein (FKRP) and LARGE may be involved in glycosylation, but their exact functions remain unknown.6 The DPM3 gene, encoding a subunit of DOL- P-Man synthase, was recently associated with CMD in a patient with muscular dystrophy and reduced α-dystroglycan staining but without intellectual disabilities.9 Finally, mutations in DAG1 encoding α-dystroglycan were identified in a patient with a form of limb girdle muscular dystrophy who had normal brain imaging and relatively mild muscular involvement but intellectual disabilities.10
POMT2 mutations have been associated with a wide spectrum of phenotypes, including some patients without structural CNS involvement and a more benign course of cognitive impairment and microcephaly.2 Other more severe forms include structural brain abnormalities such as seen in a WWS-like phenotype,11 a muscle-eye-brain phenotype,6 and cerebellar hypoplasia with or without cerebral atrophy.3
The specific mutant allele found in our patients (Y666C) has previously been reported in the homozygous state in families of Spanish and French origin and appears to be a European founder mutation based on the identification of a single haplotype (PMID: 17634419). The neuromuscular features of these other patients overlap with those found in our patients, although the authors of the previous report describe left ventricular hypertrophy in one patient as the only recognized cardiac manifestation.
The neurologic findings in our patients include microcephaly, cognitive impairment, hypotonia, progressive weakness, scoliosis, and, in one sibling, epilepsy. One sibling additionally had a coincidental diagnosis of neurofibromatosis with a paternal history for this condition. It is not otherwise thought to be contributory to the phenotype of the described congenital muscular dystrophy. An MRI brain of patient 2 showed findings suggestive of mild deep white matter and cerebellar volume loss but with otherwise normal cerebral architecture and white matter signal (Figure 3). Beside the neurologic findings, the siblings also exhibited features often observed with secondary α-dystroglycanopathies, including elevated creatine kinase, proximal limb weakness, and joint contractures.
Secondary dystroglycanopathies are a subset of CMDs and are caused by mutations in seven different genes. Their cardiovascular phenotypes are also clinically heterogeneous. Finsterer et al. reported that cardiac involvement in dystroglycanopathies appears to be more prevalent than in other forms of CMDs and includes dilated and hypertrophic cardiomyopathy, systolic dysfunction, and myocardial fibrosis. However, our patients are the first reported to exhibit aortic disease.
In Fukuyama muscular dystrophy, cardiac involvement has been reported in six individuals from four families, each with dilated cardiomyopathy.12 Systolic dysfunction was detected in 83% of 34 individuals. Another report describes myocardial fibrosis in individuals who died from heart failure.13
Walker–Warburg syndrome is the most severe form of the dystroglycanopathies and is characterized by muscular dystrophy and severe ocular and brain malformations. Neither cardiac dysfunction nor aortic root enlargement has been described.8 Similarly, in patients diagnosed with muscle-eye-brain disease, no associated cardiac involvement has been reported.8 With regard to cardiac findings in patients with mutations in the POMT2 gene, a study of nine patients found one to have right bundle branch block.6 In a second study of four patients, one patient had left ventricular hypertrophy at the age of 3.5 years.1
The enlargement of the aortic root is usually asymptomatic and is often found incidentally in the course of imaging studies. The primary causes of aortic root disease include congenital, genetic, degenerative, mechanical, inflammatory, and infectious processes that damage the aorta in a variety of ways.14 The exact mechanism of the aortopathy seen in our cohort is not clear.
Although aortopathy has not previously been reported in patients with α-dystroglycanopathies, medical therapy in the form of atenolol was initiated consistent with accepted guidelines to decrease aortic wall stress and slow progression of disease. The use of medical therapy was pursued for treatment of LV systolic dysfunction consistent with accepted guidelines. Similar medical approaches were pursued in both of the younger siblings in an attempt to improve myocardial function and slow progression of aortic dilatation.
Our results confirm previous reports that POMT2 mutations can result in a wide spectrum of phenotypes.8 However, to the best of our knowledge, the literature contains no previous report of concomitant POMT2 mutations associated with enlargement of the aortic root. Our patients demonstrate that both aortic disease and LV dysfunction may progress despite the administration of medical therapy. Therefore, we suggest early noninvasive screening of the heart in patients with POMT2 mutations accompanied by appropriate institution of medical therapy along with longitudinal noninvasive imaging to assess for clinical response or progression. Echocardiography was the imaging modality used in our report that has inherent limitations in larger patients with regard to image quality and does not offer complete imaging of the thoracic aorta. We were able to reliably assess and measure the left ventricle as well the aortic root, sinotubular junction, and ascending aorta in our patients. However, advanced imaging in the form of magnetic resonance angiography (MRA) should be considered in all patients with aortopathy to assess the entire extent of the aorta and proximal branches recognizing that more diffuse involvement may be present.
There are several limitations to our study. First, we were unable to obtain any additional past medical history for the subjects. Second, the social circumstances of our patients limited our ability to perform parental analysis of the mutation to confirm that the point mutation we identified was in fact homozygous and not compound heterozygous with a deletion. However, the fact that the Y666C mutation appears to be a founder mutation and has previously been observed in the homozygous state makes an intragenic deletion less likely. Third, while intragenic and large deletions have been observed in some cases of POMT2 mutations (PMID: 19138766). Unfortunately the laboratory that carried out the diagnostic testing was unable to provide copy number analysis to exclude the possibility of an unrecognized gene deletion.
Fourth, the natural history of these brothers was limited to the 4 years of observation reported here. Fifth, advanced imaging in the form of magnetic resonance angiography (MRA) or computed tomography angiography (CTA) was not performed secondary to physical limitations precluding the ability to adequately assess the remainder of the aorta and arterial tree.
References
Yanagisawa A, Bouchet C, Van den Bergh PY et al: New POMT2 mutations causing congenital muscular dystrophy: identification of a founder mutation. Neurology 2007; 69: 1254–1260.
Messina S, Mora M, Pegoraro et al: POMT1 and POMT2 mutations in CMD patients: a multicentric Italian study. Neuromuscul Disord 2008; 18: 565–571.
van Reeuwijk J, Brunner HG, van Bokhoven H et al: Glyc-O-genetics of Walker-Warburg syndrome. Clin Genet 2005; 67: 281–289.
Willer T, Amselgruber W, Deutzmann R, Strahl S et al: Characterization of POMT2, a novel member of the PMT protein O- mannosyltransferase family specifically localized to the acrosome of mammalian spermatids. Glycobiology 2002; 12: 771–783.
Manya H, Chiba A, Yoshida A et al: Demonstration of mammalian protein O-mannosyltransferase activity: coexpression of POMT1 and POMT2 required for enzymatic activity. Proc Natl Acad Sci USA 2004; 101: 500–505.
Godfrey C, Clement E, Mein et al: Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 2007; 130: 2725–2735.
Prados B, Pena A, Cotarelo RP, Valero MC, Cruces J : Expression of the murine Pomt1 gene in both the developing brain and adult muscle tissues and its relationship with clinical aspects of Walker-Warburg syndrome. Am J Pathol 2007; 170: 1659–1668.
Finsterer J, Ramaciotti C, Wang CH et al: Cardiac findings in congenital muscular dystrophies. Pediatrics 2010; 126: 538–545.
Lefeber D, Schönberger J, Morava E, Guillard M, Huyben K, Verrijp K et al: Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Am J Hum Genet 2009; 85: 76–86.
Hara Y, Balci-Hayta B, Yoshida-Moriguchi T, Kanagawa M et al: A dystroglycan mutation associated with limb girdle muscular dystrophy. N Engl J Med 2011; 364: 939–946.
van Reeuwijk J, Janssen M, van den Elzen C et al: POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J Med Genet 2005; 42: 907–912.
Murakami T, Hayashi YK, Noguchi et al: Fukutin gene mutations cause dilated cardiomyopathy with minimal muscle weakness. Ann Neurol 2006; 60: 597–602.
Nakanishi T, Sakauchi M, Kaneda Y et al: Cardiac involvement in Fukuyama-type congenital muscular dystrophy. Pediatrics 2006; 117: e1187–e1192.
Hiratzka LF, Bakris GL, Beckman et al: ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the diagnosis and management of patients with thoracic aortic disease. A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. J Am Coll Cardiol 2010; 55: e27–e129.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Martinez, H., Craigen, W., Ummat, M. et al. Novel cardiovascular findings in association with a POMT2 mutation: three siblings with α-dystroglycanopathy. Eur J Hum Genet 22, 486–491 (2014). https://doi.org/10.1038/ejhg.2013.165
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.165
Keywords
This article is cited by
-
Cardiac complications of congenital disorders of glycosylation (CDG): a systematic review of the literature
Journal of Inherited Metabolic Disease (2017)
