
Abstract
Socioeconomic and cultural factors are thought to have an important role in influencing human population genetic structure. To explain such population structure differences, most studies analyse genetic differences among widely dispersed human populations. In contrast, we have studied the genetic structure of an ethnic group occupying a single village in north-eastern Ghana. We found a markedly skewed male population substructure because of an almost complete lack of male gene flow among Bimoba clans in this village. We also observed a deep male substructure within one of the clans in this village. Among all males, we observed only three Y-single-nucleotide polymorphism (SNP) haplogroups: E1b1a*-M2, E1b1a7a*-U174 and E1b1a8a*-U209, P277, P278. In contrast to the marked Y-chromosomal substructure, mitochondrial DNA HVS-1 sequence variation and autosomal short-tandem repeats variation patterns indicate high genetic diversities and a virtually random female-mediated gene flow among clans. On the extreme micro-geographical scale of this single Bimoba village, correspondence between the Y-chromosome lineages and clan membership could be due to the combined effects of the strict patrilocal and patrilineal structure. If translated to larger geographic scales, our results would imply that the extent of variation in uniparentally inherited genetic markers, which are typically associated with historical migration on a continental scale, could equally likely be the result of many small and different cumulative effects of social factors such as clan membership that act at a local scale. Such local scale effects should therefore be considered in genetic studies, especially those that use uniparental markers, before making inferences about human history at large.
Similar content being viewed by others


Introduction
The genetic structure of human populations is strongly shaped by the social, cultural and demographic processes that govern migration and settlement of individuals.1, 2 Such processes are presumed to act on a number of different geographical scales, varying from a local scale – for example, within a single village, a more regional scale – over distances of a few 100 km – to sub-continental scale, reflecting distances of many hundreds to thousands of kilometres. Exactly how these processes operate on a local scale, and how these local effects influence the genetic patterns that we see on regional or sub-continental scale has not been tested extensively. Nevertheless, genetic variation over large geographic scales has been routinely investigated to infer the relationships among populations in historical contexts. The frequently observed clinal pattern of reduced genetic diversity away from Africa is seen as strong evidence for the out-of-Africa movement(s) of anatomically modern humans.3 On a sub-continental scale, the demographic changes that are inferred from genetic data are still hotly debated for Oceania,4 Europe5 and the Americas.6 Within Africa south of the Sahara, such studies are now also emerging slowly,7, 8, 9 mainly because the distribution and size of samples are limited. Most population genetic research in Africa has focused on the complex expansion patterns of Bantu-speaking people from central Africa to the south, which seems to have left strong signals in uniparental and autosomal markers across the genome.3 For West Africa, however, there are some detailed genetic studies on smaller scales.9, 10, 11, 12, 13, 14 For instance, Coelho et al13 reveal strong patterns in the genetic structure of human populations on the small island of Sao Tomé that were influenced by spatial- and temporal-specific events. Ottoni et al15 show strong founder effects and drift that have resulted in very different paternal lineages in two Libyan villages. Barbieri et al13 found a clear structure in the paternal line that matches linguistic affiliation across ethnolinguistic groups in Burkina Faso. In contrast, Veeramah et al12 show that there is little genetic structure among neighbouring ethnic groups in southern Nigeria despite the strong language differentiation among them. Apparently, this region has a particularly high diversity of ethnicities, languages, and subsistence modes, and a complex correspondence with uniparental genetic variation.
For long, anthropologists have recognized problems with too simplistic interpretations of patterns of genetic variation at a macro-scale and called for more local studies.16, 17 We consider three reasons to investigate social–cultural factors in relation to genetic studies on local, regional or sub-continental scale in West Africa. First, based on oral tradition, many Africans claim major migration events in their ancestry.18, 19, 20 Genes have been on the move within Africa on a large scale for probably a long time, for highly variable reasons.21, 22 Therefore, to better understand African migration history from a genetic point of view, we need a much denser geographical sampling. Second, the role of social factors in asymmetric gene flow between sexes has been recognized before. On a continental and sub-continental scale, this can be explained by sex-biased rates of admixture,23 subsistence mode and marital residence patterns; a general consensus is the higher female to male migration rate in Africa south of the Sahara.23, 24, 25, 26 Third, population labels (ethnicity, language and geographic ‘origin’) are often used and understood to be rigid entities, but it is widely recognized that in many cases these group labels are very flexible and influenced by social factors and distance.27
We take a next step towards a detailed, local scale genetic study and describe the results of a genetic survey among 205 males from a single Bimoba village in the Garu-Tempane district in Upper East Region of Ghana, Africa. In this area, Leiden University Medical Center was involved in medical genetic research projects since 2004.28, 29, 30, 31 We investigate the population genetic structure in this region to better inform a number of genetic association studies. There is very little known about the origins of the Bimoba. Most sources state that the Bimoba, as a tribe, represent a combination of a variable number of smaller groups.32 They are closely related to the Moba from neighbouring areas in Togo.33 The Bimoba speak Moba, which is part of the Gur language group (Niger-Congo family).9 In contrast to surrounding tribes, the Bimoba currently belong to the acephalous tribes, that is, there are no kings, chiefs or big men.34 Among the Bimoba, clan and clan group are the social focal points. These clans are patrilineally organized. All males in the village studied belong to six different self-identified clans: Baakpang, Tont, Miir, Sisiak, Najakbab and Nabakib. Although history varies from clan to clan, they all share the history of their first chief, Turiŋme. When the Bimoba settled in north-eastern Ghana and western Togo, they occupied the least fertile and most remote parts of this region, mainly in the area they still live in. This suggests that they were not able to, or did not want to rival the existing political forces at large. As a result, the Bimoba still are a group with limited power in this region. As most populations in the Sahel region, the Bimoba are pastoral agriculturalists. Both animal husbandry and crop fields are important for subsistence. The Bimoba are polygynous and practice clan exogamy, that is, within-clan marriage is prohibited. Marriage divorce is possible, but not common. Meij et al30 provides a more detailed anthropological description of the Bimoba of north-eastern Ghana.
The males in our sample were genetically screened for a set of 15 autosomal short-tandem repeats (STRs), 15 Y-chromosomal STRs and for 65 biallelic Y-chromosomal single-nucleotide polymorphisms (SNPs) defining Y-haplogroup E and sub-lineages thereof.35 In addition, a 365-bp sequence in HVS1 of their mitochondrial DNA (mtDNA) was sequenced.
Materials and methods
Research area and population background
The study area is located in the upper east region of Ghana, between 0.226W–10.689N and 0.81W–10.837N. Within this study area of approximately 360 km2, there are about 23 000 inhabitants living in over 1200 individual compounds, which are clustered in 24 villages. The upper east region of Ghana is an area with little development towards a modern industrialized society. Most of the inhabitants are traditional agriculturalists. People live in family compounds, which are essentially small farms that produce at subsistence level. The population has a patrilocal and patrilineal structure: the women are accepted to their husbands’ clan and the males stay in or around their fathers’ compound. It is custom not to marry inside one’s clan (clan-exogamy) and polygyny is widespread.
As there are no civil registries in the region, GPS coordinates of all villages and compounds within the study area were registered and assigned a unique identification number.29 The name, sex, age and tribe of each individual were collected from interviews during field visits to each household. We interviewed the head of the household (landlord) about the ethnic group and the clan of each individual. In addition, we interviewed the elders of the village from different clans on their male ancestors. The interviews were taken by a staff member from The Netherlands together with a translator enroled in the project. The translator was a lifelong inhabitant of the village under study. The demographic information from these interviews is continuously checked during the annual follow-up by revisiting all households, thereby also registering individuals that were newly born, deceased, or migrated. We also performed random, independent double household visits and this has shown that the database is accurate and reliable.
Sampling procedure and ethical approval
For the purpose of this study, we concentrated on a single Bimoba village. Genetic data were obtained from 205 men living in 93 compounds (Figure 1). The inclusion criterion was to sample at least one male from each compound in the village, two males were preferred where available, and closely related individuals such as father–son pairs were randomly included. In this village, members of the following six different Bimoba clans were sampled: Baakpang (n=90), Tont (n=43), Miir (n=55), Sisiak (n=3), Najakpab (n=8) and Nabakib (n=6). Baakpang and Tont clans claim common ancestry, similarly to Miir and Sisiak. Biological material was collected using buccal swabs. DNA was isolated from buccal swab samples using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany), according to the manufacturers’ standard protocol. This research project was executed with the informed consent of participating individuals, and approved by the ethical committees of the Ghana Health Service and the Leiden University Medical Center.

The geographical distribution of compounds (circles) in the village. Compounds are colour-labelled according to: (a) the Y-sub-haplogroup within Y-E1b1a*-M2, (b) the reported clan. The lines correspond to the spatial classification of the compound labels with a density-based Parzen classifier. The diameter of each circle is proportional to the number of sampled males in that compound; the smallest diameter represents one male, the largest represents nine males. The colour coding in (b) corresponds to that used in Figure 2.
Genotyping of autosomal and Y-chromosomal microsatellite loci
We used the Powerplex16 System amplification kit (Promega, Fitchburg, WI, USA) for 15 autosomal STR loci and the Amelogenin locus for gender identification. The AmpFℓSTRYfiler PCR Amplification Kit (Life Technologies, Carlsbad, CA, US) was used for 16 Y-chromosomal STR loci. PCR reactions were performed according to the manufacturer’s manual specifications. PCR products were analysed using an ABI 3100 automated DNA sequencer and the GenemapperID software (Life Technologies). Y-STR data can be found in Supplementary Information, Table S1.
MtDNA sequence analysis
We sequenced a fragment of 365 bp of mtDNA HVS1 (between positions 16 024 and 16 389) relative to rCRS36 essentially as described in Gabriel et al.37 Sequenced fragments were analysed on an ABI 3100 automated DNA sequencer and the SeqScape software (Life Technologies). Sequences were manually aligned and edited using BioEdit vs. 7.0.5.2.38 Before analysis, the 10-bp C-stretch (between 16 084 and 16 093) was removed from the aligned sequences.
Genotyping of Y-SNP polymorphisms
A total of 65 haplogroup-informative Y-chromosome SNPs were typed using the multiplex SNaPshot method (Life Technologies). We performed a stepwise analysis using different primer mixes for different levels in the phylogeny (Supplementary Information, Table S2). In a first experiment, we typed SNPs specific for the main Y-haplogroups (Supplementary Information, Figure S1). Subsequently, we typed SNPs that specify most known E sub-haplogroups. In the final step, we typed the SNPs that further differentiate sub-haplogroup E1b1a*-M2). An additional SNP, V39,39 was sequenced on the PGM semiconductor sequencer using the IonXpress Plus Fragment Library Preparation Kit (Life Technologies). The methods and protocols are described in detail in the Supplementary Information.
Spatial classification analysis
In order to formally test for non-randomness in the geographical separation of haplogroups and clans, we used a supervised, density-based Parzen classifier.40 Parzen classification is a technique for nonparametric density estimation, which can also be used for classification. It can be regarded as a generalization of k-nearest neighbour techniques. However, rather than choosing only the k nearest neighbours of a test point and labelling the test point with the weighted majority of its neighbours’ values, one can consider all points simultaneously. In the context of this paper, Parzen classifier estimates the densities of each clan or Y-haplogroup category by evaluating the distance-weighed contributions of each compound. The distance over which the contribution is evaluated is defined by sliding window function. The window size was optimized for low classification error. The classification error of the Parzen classifier has been estimated by a leave one out (LOO) procedure.40, 41, 42 A LOO procedure estimates the error by training the classifier on all but one individual; this individual is used for testing. Iteration allows the evaluating of the effect of exclusion of every individual in the data set and the average of the resulting error is an unbiased evaluator of classification. We used the Parzen classifier implemented in the PR-tools43 for Matlab CO package. T-test analysis was performed in the standard Matlab CO package (Matlab, Natick, MA, USA). Parzen classifier parzenc function was used for the computation of the optimum smoothing parameter between classes.43 For the LOO procedure,44 we used the random label reshuffling crossval function in PR-tools. Rand45 evaluation of the correspondence of classifications has been implemented by the authors as a Matlab routine, and is available on request.
Statistical analyses
We used Arlequin version 3.11 to perform analysis of molecular variance (AMOVA)46, 47 among the clans and to estimate gene diversities within clans for each different genetic system tested. We used Network48 version 4.2.0.1 to draw median joining networks based on the combined Y-STR and Y-SNP information, and on HVS1 sequence variation. For both genetic systems, a variable weight was given to different variable loci. In order to estimate these different weights, we first drew a network giving all positions an equal weight and used the statistics option on the fully drawn network to obtain an estimate of the rate of homoplasy. Based on these estimates, highly homoplasic positions were down weighed accordingly.
Results
Spatial distribution of the clans and Y-haplogroups
We have investigated the spatial distribution of Y-haplogroups (Figure 1a) and clans (Figure 1b) within the village and we have observed a distinct non-random distribution of both. In order to formally test the geographical separation of haplogroups and clans, we used a supervised, density-based Parzen classifier.40 Using this method, we were able to geographically separate haplogroups into clusters of same clan and haplogroup (Figure 1a). Clan classification had an error of 3.92% and Y-haplogroup classification had an error of 6.86%. These values indicate that for any single individual his haplogroup can be established on basis of his neighbours with a classification certainty of 93.14% (Figure 1a). The clan can be established with 96.08% certainty (Figure 1b). In addition, we compared the observed classifier performance with the clustering of the compounds with randomly shuffled labels (100 trials). The Student’s t-test rejected the null-hypothesis of random spatial clustering with P-values of 4.53*10−49 and 8.23*10−92 for Y-haplogroups and clans, respectively. Thus, spatial clustering of Y-haplogroups and clans are both significantly non-random. The higher classification error of Y-haplogroups compared with clans is caused by the inability to geographically separate males carrying different Y-haplogroups that live in the same compound.
Second, we examined the correspondence between haplogroup clusters and clan clusters. In order to test whether compounds that harbour the same clan are also more likely to contain the same Y-haplogroup, we used a W Rand similarity measure.45 This statistic measures to what degree two classifications match each other. Self-reported clan membership and Y-haplogroups have W Rand index of 0.69, which means that the Y-haplogroup can be correctly identified from the clan membership for 69% of the compounds.
Genetic diversity in the village
Next, we studied the genetic variation within and among the different clans. For this we estimated within clan and among clan genetic variation by means of FST, AMOVA46, 47, 49 and gene diversity across autosomal STR, Y-STR and HVS1 loci. The results of these analyses are shown in Tables 1 and 2. Y-STR data can be found in Supplementary Information, Table S1. There are highly significant Y-chromosomal genetic differences among the clans, but the clans are not significantly differentiated when analysing mtDNA HVS1 genetic variation. This is also expressed by the very high among-population (ie, among clan) genetic variance (59.9%) observed for Y-STRs, compared with the very low estimates for autosomal STRs (1.19%) and HVS1 (0.28%; Table 2). Our result strongly suggests a very reduced – if not absent – male-mediated gene flow among the clans and a random female-mediated gene flow among the same clans.
Detailed Y-chromosome and mtDNA variation among clans: haplotypes and haplogroups
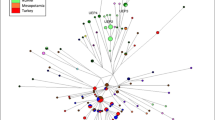
The marked gender-specific difference in gene flow among the clans is also reflected in the distribution of the Y-STR haplotypes and HVS1 sequence haplotypes among the clans (Figure 2a). It is further confirmed by analysing the clan-specific Y-haplogroup distribution (Table 3). Most of males in this village belong to only three Y-haplogroups E1b1a*-M2, E1b1a7a*-U174 and E1b1a8a*-U209, P277, P278 (Figure 1a). This distribution is significantly non-random (P<0.001, Monte–Carlo Fisher’s exact test). Except for members of the largest clan (Baakpang), most members of the other five clans display Y-STRs belonging to a single Y-haplogroup, and appear strongly clustered in the Y-STR network. Such a clear clustering is not observed in the HVS1 network (Figure 2b). The Y-related clan-specific correspondence is also obvious when plotting and combining the Y-haplogroup distribution (Figure 1a) with the clan-specific distribution (Figure 1b) across compounds.

Median joining networks for (a) Y-STR haplotypes, and (b) mtDNA HVS1 sequence haplotypes. Each haplotype pie is colour-labelled according to the reported clan of males carrying that haplotype; the colour labels correspond to those in Figure 1b. In the Y-STR haplotype network (a), the segments with the same Y-haplogroup are indicated by a similar background colour that corresponds to the colour labels in Figure 1a. The smallest distance between two haplotypes equals one repeat length difference in one STR. The diameter of each pie is proportional to the frequency of that haplotype; the smallest pies represent a single male, the largest 81 males.
Discussion
In our study area, a single Bimoba village in the north-east of Ghana, the Y-chromosomal genetic structure is correlated with the spatial distribution of the compounds in the village. The spatial Parzen classification scores for both clans and Y-haplogroups indicate that the clan identity and genetic lineage of a male in one compound can be reliably estimated by those of neighbouring compounds. Such a settlement pattern can be explained by patrilocal settlement (Figure 1b). Y-chromosome lineages settle distantly only when there is a land-shortage around a core clan area. This suggests a direct relation of land inheritance with agricultural life style.
Our study shows that the role of an important social factor in shaping genetic structure appears to be at play on a much more local scale than previously thought and tested. It is known for quite some time that there are significant gender-specific differences in a number of demographic processes among populations in Africa,7, 23, 25 and elsewhere.50, 51 Such differences were found to depend strongly on the social structure. Among traditional hunter–gatherer populations, such as central African pygmies, female-mediated gene flow (as detected by mtDNA variation) is substantially reduced compared with male-mediated gene flow (as detected by Y-chromosomal genetic variation patterns).25 Exactly the reverse is generally observed among traditional farming communities. This is usually attributed to the combined influence of patrilocality and polygyny, which appears to be the dominant type of social structure among many African farming groups,25 like in the Bimoba.
We found a markedly skewed male population substructure because of an almost complete lack of male gene flow among clans in a single Bimoba village. In contrast, female gene flow was not confined to clans; evidence of clan exogamy reportedly practiced by Bimoba. Based on an anthropological study33 and on our observations in the field,30 we know that clan structure has a vital role in many cultural and demographic aspects of daily life among the Bimoba. It is remarkable that one clan is composed of two distinct Y-haplogroup lineages, whereas the other clans are homogeneous in their Y-haplogroup composition (Figure 2a). This deviation, from the otherwise strict genetically defined group membership, indicates the importance of clan as a social factor.
Implications
The pattern of genetic diversity and gene flow in Y-chromosomes that we find is as strong and deep as can be found on a much larger geographical scale throughout West Africa.9, 12 The three Y-haplogroups present in this village are found at high, but varying, frequencies across Africa.9 Two Y-haplogroups E1b1a7a*-U174 and E1b1a8a*-U209, P277, P278 dominate in groups of Bantu-speaking males across central and southern Africa.9 The other haplogroup, E1b1a*-M2, is more frequent towards the west of Africa.9 We do not find haplotype sharing among the three haplogroups, which indicates that these lineages are rooted deeply and distinctly in the E1b1a*-M2 lineage. Our result of clan specific Y-chromosomal lineages together with results of de Filippo et al9 indicate a scenario where several patrilineally related genetic lineages constitute a ‘tribe’. The fact that we find similar patrilineal lineages across West Africa10 raises the intriguing possibility that many ethnic groups (or ‘tribes’) or even ethnolinguistic groups14 that identify themselves as homogenously related entities, consist of the same small number of closely related genetic lineages. The consequence is that the relative frequency of these Y-chromosomal lineages (i.e., the Y-haplogroups) within such constructed groups could be caused entirely by the relative contributions of distinct clans within an ethnic group. Such contributions are probably highly stochastic (cf. Ottoni et al15). Additional study of this village, as well as collecting similar data from more villages and in different tribes in the same region is ongoing and will further improve our knowledge of these people and the factors that shape their Y-chromosomal population structure.
Conclusion
On the geographic scale of a single village, population genetic structure among the traditional agricultural people of the Bimoba is strongly influenced by social structures. There is a highly skewed male population substructure, caused by clan membership. Female-mediated gene flow seems random. This genetic pattern can be explained by the patrilocal and patrilineal structure in such societies. Clearly, the role of local social factors needs to be considered across large parts of the continent. There is an explicit assumption in population genetic studies that more widespread sampling will reveal more ancient demographic patterns, and thus local sampling will reveal only the most recent demographic events.26 Our results indicate this need not be true: what is perceived as genetic structure because of geography or language variation on a large scale can in fact be explained equally well by social factors acting on local scale. Future sampling efforts should consider this.
References
Cavalli-Sforza LL, Menozzi P, Piazza A : The History and Geography of Human Genes. Princeton Yniversity Press, 1994.
Curran SR, Agardy T : Common property systems, migration, and coastal ecosystems. J Human Environ 2002; 31: 303–305.
Barbujani G, Colonna V : Human genome diversity: frequently asked questions. Trends Genet 2010; 26: 285–295.
Wollstein A, Lao O, Becker C et al: Demographic history of Oceania inferred from genome-wide data. Curr Biol 2010; 20: 1983–1992.
Haak W, Balanovsky O, Sanchez JJ et al: Ancient DNA from European early neolithic farmers reveals their near eastern affinities. PLoS Biol 2010; 8: e1000536.
O'Rourke DH, Raff JA : The human genetic history of the Americas: the final frontier. Curr Biol 2010; 20: R202–R207.
Tishkoff SA, Williams SM : Genetic analysis of African populations: human evolution and complex disease. Nat Rev Genet 2002; 3: 611–621.
Adeyemo AA, Chen G, Chen Y, Rotimi C : Genetic structure in four West African population groups. BMC Genet 2005; 6: 38.
de Filippo C, Barbieri C, Whitten M et al: Y-chromosomal variation in Sub-Saharan Africa: insights into the history of Niger-Congo groups. Mol Biol Evol 2011; 28: 1255–1269.
Campbell MC, Tishkoff SA : The evolution of human genetic and phenotypic variation in Africa. Curr Biol 2010; 20: R166–R173.
Rosa A, Ornelas C, Jobling MA, Brehm A, Villems R : Y-chromosomal diversity in the population of Guinea-Bissau: a multiethnic perspective. BMC Evol Biol 2007; 7: 124.
Veeramah KR, Connell BA, Pour NA et al: Little genetic differentiation as assessed by uniparental markers in the presence of substantial language variation in peoples of the Cross River region of Nigeria. BMC Evol Biol 2010; 10: 92.
Coelho M, Coia Cintia Alves V, Luiselli D et al: Human microevolution and the Atlantic slave trade: a case study from Sao Tome. Curr Anthropol 2008; 49: 134–143.
Barbieri C, Whitten M, Beyer K, Schreiber H, Li M, Pakendorf B : Contrasting maternal and paternal histories in the linguistic context of Burkina Faso. Mol Biol Evol 2012; 29: 1213–1236.
Ottoni C, Larmuseau MH, Vanderheyden N et al: Deep into the roots of the Libyan Tuareg: a genetic survey of their paternal heritage. Am J Phys Anthropol 2011; 145: 118–124.
MacEachern S : Genes, tribes, and African history. Curr Anthropol 2000; 41: 357–384.
Keita SOY, Kittles RA : The persistence of racial thinking and the myth of racial divergence. Am Anthropologist 1997; 99: 534–544.
France P : The Kaunitoni migration: notes on the genesis of a Fijian tradition. J Pacific History 1966; 1: 107–113.
Stevens P : The Kisra legend and the distortion of historical tradition. J African History 1975; 16: 185–200.
Rippen BV : Practices and customs of the African natives involving dental procedure. Am Anthropologist 1918; 20: 461–463.
Baker J, Aina TA : The Migration Experience in Africa. Uppsala: Nordiska Afrikainstitutet, 1995.
Ki-Zerbo J, Unesco: International scientific committee for the drafting of a general history of A. General history of Africa. 1, Methodology and African prehistory. London; Berkeley; Paris: Heinemann; University of California Press; Unesco, 1981.
Wood ET, Stover DA, Ehret C et al: Contrasting patterns of Y chromosome and mtDNA variation in Africa: evidence for sex-biased demographic processes. Eur J Hum Genet 2005; 13: 867–876.
Destro-Bisol G : Genetic variation and social structure: a case-study from Africa. Human Evolution 2005; 20: 93–98.
Destro-Bisol G, Donati F, Coia V et al: Variation of female and male lineages in sub-Saharan populations: the importance of sociocultural factors. Mol Biol Evol 2004; 21: 1673–1682.
Wilkins JF, Marlowe FW : Sex-biased migration in humans: what should we expect from genetic data? Mol Cell Develop Biol 2006; 28: 290–300.
Zeleza PT : The inventions of African identities and languages: the discursive and developmental implications. Selected Proceedings of the 36th Annual Conference on African Linguistics Georgia Southern University, Statesboro Georgia, USA 2006.
Gasser RB, de Gruijter JM, Polderman AM : Insights into the epidemiology and genetic make-up of Oesophagostomum bifurcum from human and non-human primates using molecular tools. Parasitology 2006; 132: 453–460.
Ziem JB, Olsen A, Magnussen P et al: Distribution and clustering of Oesophagostomum bifurcum and hookworm infections in northern Ghana. Parasitology 2006; 132: 525–534.
Meij JJ Testing life history theory in a contemporary African population. Department of Geriatrics and Gerontology, Faculty of Medicine, Leiden University Medical Center (LUMC), Leiden University 2008.
Meij JJ, Van Bodegom D, Ziem JB et al: Quality–quantity trade-off of human offspring under adverse environmental conditions. J Evol Biol 2009; 22: 1014–1023.
Assimeng M : Bimoba Sociological Study. Legon: University of Ghana, Legon, 1990.
Zwernemann J : Studien zur Kultur der Moba (Nord-Togo). Köppe, 1998.
Middleton J, Tait D : Tribes Without Rulers: Studies in African Segmentary Systems. London: Routledge & Paul, 1958.
Karafet TM : New binary polymorphisms reshape and increase resolution of the human Y chromosomal haplogroup tree. Genome Res 2008; 18: 830.
Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N : Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet 1999; 23: 147.
Gabriel MN, Huffine EF, Ryan JH, Holland MM, Parsons TJ : Improved MtDNA sequence analysis of forensic remains using a ‘mini-primer set’ amplification strategy. J Forensic Sci 2001; 46: 247–253.
Hall T : BioEdit. Carlsbad, CA, USA: Ibis Biosciences.
Trombetta B, Cruciani F, Sellitto D, Scozzari R : A new topology of the human Y chromosome haplogroup E1b1 (E-P2) revealed through the use of newly characterized binary polymorphisms. PLoS One 2011; 6: e16073.
Parzen E : On estimation of a probability density function and mode. Ann Mathematical Stat 1962; 33: 1065–1076.
Lissack T, Fu KS : Error estimation in pattern recognition viaL_alpha-distance between posterior density functions. IEEE Transactions on Information Theory 1976; 22: 34–45.
Van der Heijden F, Duin RPW, De Ridder D, Tax D : Classification, Parameter Estimation and State Estimation. Wiley Online Library, 2004.
Duin PJ, Paclik P, Pekalska E, de Ridder D, Tax DMJ, Verzakov S : PRTools4.1, A Matlab Toolbox for Pattern Recognition. Delft University of Technology, 2007.
Kohavi R A study of cross validation and bootstrap for accuracy estimation and model selection. International Joint Conference on Artificial Intelligence IJCAI 1995.
Rand WM : Objective criteria for the evaluation of clustering methods. J Am Stat Assoc 1971; 66: 846–884.
Excoffier L, Laval G, Schneider S : Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evol Bioinform Online 2007; 1: 47–50.
Excoffier L, Smouse PE, Quattro JM : Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 1992; 131: 479–491.
Bandelt HJ, Forster P, Rohl A : Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 1999; 16: 37–48.
Meirmans PG : Using the AMOVA framework to estimate a standardized genetic differentiation measure. Evolution 2006; 60: 2399–2402.
Chaix R, Quintana-Murci L, Hegay T et al: From social to genetic structures in central Asia. Curr Biol 2007; 17: 43–48.
Gunnarsdottir ED, Nandineni MR, Li M et al: Larger mitochondrial DNA than Y-chromosome differences between matrilocal and patrilocal groups from Sumatra. Nat Commun 2011; 2: 228.
Acknowledgements
We thank the members of the Garu-Tempane district, Upper East Region, Ghana and the Ghana field staff for their enthusiasm and kind participation and hospitality during our fieldwork, and David van Bodegom and Ulrika Eriksson for their advice and comments. This study was supported by an unrestricted grant of the Board of the Leiden University Medical Center, Leiden, The Netherlands, and the EU Network of Excellence ‘LifeSpan’ (EU FP6 036894).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Sanchez-Faddeev, H., Pijpe, J., van der Hulle, T. et al. The influence of clan structure on the genetic variation in a single Ghanaian village. Eur J Hum Genet 21, 1134–1139 (2013). https://doi.org/10.1038/ejhg.2013.12
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.12
Keywords
This article is cited by
-
Ecological effects and perceptions of victims of pipeline explosion in a developing country
International Journal of Environmental Science and Technology (2015)
