
Abstract
Congenital nystagmus (NYS) is characterized by bilateral, spontaneous, and involuntary movements of the eyeballs that most commonly presents between 2 and 6 months of life. To date, 44 different FRMD7 gene mutations have been found to be etiological factors for the NYS1 locus at Xq26-q27. The aim of this study was to find the FRMD7 gene mutations in a large eleven-generation Indian pedigree with 71 members who are affected by NYS. Mutation analysis of the entire coding region and splice junctions of the FRMD7 gene revealed a novel missense mutation, c.A917G, predicts a substitution of Arg for Gln at codon 305 (Q305R) within exon 10 of FRMD7. The mutation was detected in hemizygous males, and in homozygous and heterozygous states in affected female members of the family. This mutation was not detected in unaffected members of the family or in 100 unrelated control subjects. This mutation was found to be at a highly conserved residue within the FERM-adjacent domain in affected members of the family. Structure prediction and energetic analysis of wild-type FRMD7 compared with mutant (Q305R) revealed that this change in amino acid led to a change in secondary structure predicted to be an energetically unstable protein. The present study represents the first confirmation of FRMD7 gene mutations in a multigenerational Indian family and expands the mutation spectrum for this locus.
Similar content being viewed by others
Introduction
Nystagmus (NYS), one of the most common neuro-ophthalmological disorders among live births, often seriously reduces visual acuity.1 NYS could be physiological or pathological and the latter can be divided into acquired or congenital forms. Although congenital NYS is essentially present at birth, it can also occur early in the child's vision development. The estimated incidence of NYS in the general population was 2.4/1000.2 Most cases of early onset NYS are hereditary and are transmitted in X-linked dominant, X-linked recessive, and autosomal dominant forms with incomplete penetrance and variable expressivity. Several family-based, genome-wide linkage studies showed evidence of linkage at seven NYS loci on Xq26.2 (NYS1 (MIM 31 0700 )), 6p12 (NYS2 (MIM)), 7p11.2 (NYS3 (MIM 60 8345 )), 13q31-q33 (NYS4 (MIM 19 3003) ), Xp11.4 (NYS5 (MIM 30 0589 )), Xp22.3 (NYS6 (MIM 30 0814 )) and on 1q31-q32.2.3
We have analyzed a large consanguineous Indian family diagnosed as having NYS. Sequencing of the FRMD7 gene, which is well established in its involvement in the pathogenesis of X-linked NYS, revealed a novel missense mutation, c.A917G, that predicts a substitution of Arg for Gln at codon 305 (Q305R) within exon 10 in the affected members of this family.
Methods
Family Studies
This large eleven-generation pedigree (UR031) exhibiting isolated non-syndromic congenital NYS is from southern India. Experienced ophthalmologists (RS) performed detailed examinations on the 36 selected patients, testing visual acuities, slit lamp, color vision, intraocular pressure, transillumination defects of the iris, ocular oscillations, fundoscopy and performing visual evoked potentials as well. Angle of head turn was measured with the help of deviometer and electroretinogram testing was performed in selected affecteds.
Mutation analysis
Genomic DNA from 29 family members and 100 normal controls were PCR amplified for regions covering the entire coding sequence and splice junctions of the FRMD7 gene. Detailed methodology is described in Supplementary Methods.
Protein modeling
The secondary structure of human wild-type and mutant FRMD7 was predicted using I-TASSER,4 and the results saved in PDB file format. The PDB files obtained for the two samples were then used by Jmol to visualize the structures of these proteins.5 The effect of amino-acid replacement on structure or function was predicted by the PolyPhen-2 program,6 which calculates position-specific independent counts and computes the difference between profile scores of both allelic variants in the polymorphic position. Larger positive values of this difference are indicative that the studied substitution is rarely or never observed in the protein family. The model for evaluating rare alleles, dense mapping of regions identified by genome-wide association studies, and analysis of natural selection were used to obtain the confidence score. Finally, the energy profiles were generated using ANOLEA program to identify the likely stability of the mutant protein structure as compared with its wild type.7
Results
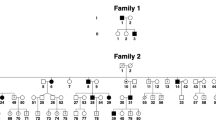
The pattern of inheritance in this 444-member family (200 males and 244 females) is consistent with X-linked dominant mode of inheritance with reduced penetrance. The age distribution of affecteds is from 6 to 90 years and onset was between 3 and 6 months of age. The family is highly consanguineous with 19 visible consanguineous marriages, and the phenotype was present in 71 members of the pedigree. The partial pedigree structure is shown in Figure 1a. All the individuals examined showed involuntary asymmetric pendular eye movements with unidirectional jerky NYS. Head tilt was observed in 20 out of 36 affecteds. No other anomalies were found apart from NYS including visual acuity, color vision, optic nerves, and retina.

(a) Partial pedigree of UR031 with NYS1. Affected individuals are shown with blackened symbols, and normal individuals are shown with clear symbols. Individuals indicated by a cross are deceased. Deceased and individuals with question mark were not examined. (b) Comparison of normal and derived amino acid sequences due to the missense mutation. (c) Conservation of amino acids is altered by missense mutations in the highly conserved residue of FERM-adjacent (FA) domain. Amino-acid sequence comparison in several related proteins. The six proteins depicted are human, Macaca mulatta, Mus musculus, Rattus norvegicus, Equus caballus, and Bos Tauru. The FRMD7 mutation is indicated above the aligned sequence, with the amino acid shaded in the alignment.
Sequence analysis of all affecteds and carriers in family (UR031) revealed a missense mutation, c.A917G, in exon 10, which would result in substitution of Arg for Gln at codon 305 (Q305R) (Figure 1b). The same mutation was not observed in unaffected males/females nor in the 100 normal controls from the same geographic region.
The structure prediction of human wild-type and mutant FRMD7 has revealed that the single nucleotide change (A>G) at position 305 results in the substitution of the amino acid glutamine (Q) in the wild type by arginine (R) in the mutant phenotype. This change in amino acid led to a change in secondary structure from helix to a coil (Figures 2a–d). Additionally, PolyPhen-2 prediction of functional effect(s) of nsSNPs showed a position-specific independent count score of 0.824. The confidence score obtained with the sensitivity value of 0.81 and specificity 0.90 predicted this mutation to be damaging to protein structure and/or function of the mutant. The stability of a protein structure can be predicted from energy calculations for each amino acid in a protein chain. If the energy of each amino acid in a given protein chain is negative, the protein is considered to be structurally stable. Based on the energy calculation using ANOLEA, a favorable energy environment would be damaged by this amino-acid substitution.

(a–f) Change in the secondary structure of FRMD7 as result of mutation of amino acid in position 305. (a) Wild type; (b) mutant; (c) residue 1–305 of wild-type protein (d): Residue 1–305 of mutant. The area indicated by the arrow head shows that the helical structure of the residue 306 is changed to coil. Mean force potential plot for the protein of human wild-type (e) and mutant (f) FRMD7. The y-axis represents the energy for each amino acid of the protein at the location given in the x-axis. The negative energy (shown in red) shows favorable energy enviornment, whereas positive energy (shown in green) represents unfavorable enviornment. A favorable energy environment tends to be damaged because of Q to R change at 305.
As the modeled protein structure of the mutant containing R in place of Q would be energetically unstable (Figures 2e–f), the mutation at position 305 changes the energy environment of the protein. The amino acids in the mutated protein tend to have positive energy values as compared with the wild-type protein, thereby disrupting the structural stability of the mutated protein.8
Discussion
FRMD7 mutations account for almost 50% of Western families with NYS and ∼5% of the sporadic cases.9 To date, 44 FRMD7 gene mutations have been identified in various NYS families suggesting that this gene has an important role in the NYS development.8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 The majority of these mutations were clustered in the highly conserved FERM-C domain in exons of 7–9 (Table 1).
The present novel c.A917G (Q305R) mutation was observed as hemizygous in affected males (n=9), and homozygous (n=8) or heterozygous (n=3) in affected females. Additionally, the obligate female carriers (n=5) without disease had a heterozygous mutation. The disease was fully penetrant in all males with the mutation. All the affected females with homozygous mutation showed complete expression of the phenotype, including head tilt and NYS. As some of the obligate carrier females are not showing the phenotype, we are concluding that this could be due to incompletely penetrant dominant gene. This is consistent with a previously published report.25 The clinical significance of hemi/homozygous as compared with heterozygous mutation is not yet known, although Kaplan et al13 found a missense mutation, R229G, in the FRMD7 gene in heterozygous and homozygous condition in members of a large X-linked NYS family without phenotypic variation.
The location of the present residues is highly conserved in multiple species (Figure 1c) suggesting that the mutation at codon 305 is likely to have a significant effect on FRMD7 protein functionality. As the present reported mutation is located in a highly conserved residue of the FERM-adjacent domain and possessed similarity to other protein kinase substrates, we also hypothesized that this genetic region has regulatory functions. Further, the FERM-C domain was a mutation-rich region, suggesting that this region should be treated as the most important candidate region when screening for mutations. Prediction and energetic analysis of the structure of mutant and wild-type proteins revealed a change in the secondary structure from helix to a coil that led to an energetically unstable protein. Similarly, Li et al8 reported a predicted unstable target protein of human FRMD7 with a missense mutation (H208R) in a different location within the gene.
To our knowledge, this is the first report of homozygous, hemizygous, and heterozygous FRMD7 mutations in the same multigenerational family with isolated non-syndromic NYS with many consanguineous loops. The present data extend the mutation spectrum of FRMD7 gene mutations to a novel functional domain downstream from the FERM-C domain of the protein.
References
Spierer A : Etiology of reduced visual acuity in congenital nystagmus. Ann Ophthalmol 1991; 23: 393–397.
Sarvananthan N, Surendran M, Roberts EO et al: The prevalence of nystagmus: the Leicestershire nystagmus survey. Invest Ophthalmol Vis Sci 2009; 50: 5201–5206.
Xiao X, Li S, Guo X, Zhang Q : A novel locus for autosomal dominant congenital motor nystagmus mapped to 1q31-q32.2 between D1S2816 and D1S2692. Hum Genet 2011; 1–6, e-pub ahead of print 8 November 2011.
Roy A, Kucukural A, Zhang Y : I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 2010; 5: 725–738.
Hanson RM : Jmol - a paradigm shift in crystallographic visualization. J Appl Cryst 2010; 43: 1250–1260.
Adzhubei IA, Schmidt S, Peshkin L et al: A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Melo F, Devos D, Depiereux E, Feytmans E : ANOLEA: a www server to assess protein structures. Proc Int Conf Intell Syst Mol Biol 1997; 5: 187–190.
Li N, Wang X, Wang Y et al: Investigation of the gene mutations in two Chinese families with X-linked infantile nystagmus. Mol Vis 2011; 17: 461–468.
Tarpey P, Thomas S, Sarvananthan N et al: Mutations in FRMD7, a newly identified member of the FERM family, cause X-linked idiopathic congenital nystagmus. Nat Genet 2006; 38: 1242–1244.
Fingert JH, Roos B, Eyestone ME, Pham JD, Mellot ML, Stone E : Novel intragenic FRMD7 deletion in a pedigree with congenital X-linked nystagmus. Ophthalmic Genet 2010; 31: 77–80.
He X, Gu F, Wang Y et al: A novel mutation in FRMD7 causing X-linked idiopathic congenital nystagmus in a large family. Mol Vis 2008; 14: 56–60.
He X, Gu F, Wang Z et al: A novel frameshift mutation in FRMD7 causing X-linked idiopathic congenital nystagmus. Genet Test 2008; 12: 607–613.
Kaplan Y, Vargel I, Kansu T et al: Skewed X inactivation in an X linked nystagmus family resulted from a novel, p.R229G, missense mutation in the FRMD7 gene. Br J Ophthalmol 2008; 92: 135–141.
Li N, Wang L, Cui L et al: Five novel mutations of the FRMD7 gene in Chinese families with X-linked infantile nystagmus. Mol Vis 2008; 14: 733–738.
Li ND, Cui LH, Wang LM et al: [The G990T mutation of the FRMD7 gene in a Chinese family with congenital idiopathic nystagmus]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2008; 25: 11–14.
Li ND, Wang LM, Cui LH et al: [Study of gene mutation in a Chinese family with X-linked congenital nystagmus]. Zhonghua Yan Ke Za Zhi 2008; 44: 138–142.
Schorderet DF, Tiab L, Gaillard MC et al: Novel mutations in FRMD7 in X-linked congenital nystagmus. Mutation in brief #963. Online. Hum Mutat 2007; 28: 525.
Self JE, Shawkat F, Malpas CT et al: Allelic variation of the FRMD7 gene in congenital idiopathic nystagmus. Arch Ophthalmol 2007; 125: 1255–1263.
Shiels A, Bennett TM, Prince JB, Tychsen L : X-linked idiopathic infantile nystagmus associated with a missense mutation in FRMD7. Mol Vis 2007; 13: 2233–2241.
Zhang B, Liu Z, Zhao G : Novel human pathological mutations. Gene symbol: FRMD7. Disease: congenital motor nystagmus. Hum Genet 2007; 122: 414.
Zhang B, Liu Z, Zhao G et al: Novel mutations of the FRMD7 gene in X-linked congenital motor nystagmus. Mol Vis 2007; 13: 1674–1679.
Zhang Q, Xiao X, Li S, Guo X : FRMD7 mutations in Chinese families with X-linked congenital motor nystagmus. Mol Vis 2007; 13: 1375–1378.
Khan AO, Shinwari J, Al-Sharif L, Khalil DS, Al Tassan N : Prolonged pursuit by optokinetic drum testing in asymptomatic female carriers of novel FRMD7 splice mutation c.1050 +5 G>A. Arch Ophthalmol 2011; 129: 936–940.
Du W, Bu J, Dong J et al: A novel frame-shift mutation in FRMD7 causes X-linked idiopathic congenital nystagmus in a Chinese family. Mol Vis 2011; 17: 2765–2768.
Thomas S, Proudlock FA, Sarvananthan N et al: Phenotypical characteristics of idiopathic infantile nystagmus with and without mutations in FRMD7. Brain 2008; 131: 1259–1267.
Acknowledgements
We thank the patients for their cooperation in the study. The study was supported by Green Cross Blood Bank, Ahmedabad, India and the Swiss National Science Foundation to SEA. DB is supported by Grant Number P20 RR16469 from NCRR, component of NIH.
Web Resources
Ensembl, http://www.ensembl.org/Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for FRMD7, NYS1, NYS2, NYS3, NYS4, NYS5, and NYS6)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Radhakrishna, U., Ratnamala, U., Deutsch, S. et al. Novel homozygous, heterozygous and hemizygous FRMD7 gene mutations segregated in the same consanguineous family with congenital X-linked nystagmus. Eur J Hum Genet 20, 1032–1036 (2012). https://doi.org/10.1038/ejhg.2012.60
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.60
Keywords
This article is cited by
-
Truncated FRMD7 proteins in congenital Nystagmus: novel frameshift mutations and proteasomal pathway implications
BMC Medical Genomics (2024)
-
A novel frameshift mutation in FRMD7 causes X-linked infantile nystagmus in a Chinese family
BMC Medical Genetics (2019)
-
A FRMD7 variant in a Japanese family causes congenital nystagmus
Human Genome Variation (2015)
-
A start codon mutation of the FRMD7 gene in two Korean families with idiopathic infantile nystagmus
Scientific Reports (2015)
-
Identifcation of a Novel Mutation p.I240T in the FRMD7 gene in a Family with Congenital Nystagmus
Scientific Reports (2013)
